放射性核素在氧化物、磷酸盐和黏土矿物表面吸附:热力学和微观结构认识
2012-01-04范桥辉牛智伟许君政郭治军吴王锁
范桥辉,牛智伟,许君政,郭治军,吴王锁
兰州大学 核科学与技术学院 放射化学与核环境研究所,甘肃 兰州 730000
放射性核废物的妥善处理和安全处置是制约我国核能发展战略的关键因素之一,而放射性核素在环境介质中吸附行为的研究是放射性废物处置过程中的基础研究内容之一[1]。利用实验测得的放射性核素在固相和液相间的分配系数以及其在固相中的扩散系数,可以比较准确的预测和评估放射性核素在环境介质中的迁移行为,为核废物妥善处理和安全处置提供有效的理论指导。放射性核素在固液界面上的吸附机理主要包括表面配位作用、表面沉淀和离子交换反应等[2]。放射性核素在环境介质中的吸附行为受到多种因素控制或影响,如pH、离子强度、天然有机质、吸附质浓度和固体表面电荷性质、功能基团密度等,从而给人们对其吸附行为构建模型和探讨吸附机理带来了极大的困难。尽管大量研究工作对放射性核素在固液界面上的吸附机理进行了表观和微观上的探索和解释,但这些实验所得的模型和热力学结论的正确性或合理性主要取决于实验过程的合理与否。通过大量研究工作总结,发现通过以下7个主要步骤可得到相对准确、系统和科学的界面吸附信息:
(1)对吸附剂进行详细的结构、形貌、拓扑等物理化学性质的表征,如XRD对吸附材料的结构表征,FTIR对吸附剂表面功能基团的表征, BET或ESA法测定固体颗粒的尺寸大小和比表面积等关键性质;
(2)吸附剂材料的表面电荷性质表征,吸附剂表面电荷密度与吸附体系pH之间的关系和吸附剂表面零电荷点是影响放射性核素吸附种态、过程和机理的重要参数;
(3)通过对固体材料的表面进行连续电位滴定,研究表面可变电荷位的质子化和去质子化反应的平衡常数;
(4)开展相关的静态吸附研究,考察pH、离子强度、吸附质浓度和天然有机质等对放射性核素在固液界面吸附的影响;
(5)对放射性核素的表面吸附种态进行研究,考察放射性核素在固液界面上的微观空间结构等信息;
(6)综合以上结果得到宏观和微观结构,利用表面配位模型对放射性核素在固液界面上的吸附机理和过程进行探讨;
(7)利用先进光谱技术对所得模型和机理进行分子或原子水平上的论证。
本文拟通过几个具体实例分别对每一步骤进行详细阐述。这些实例中,主要选取兰州大学放射化学与核环境研究所针对放射性核素在固液界面吸附的研究成果,涉及放射性核素在氧化物(如氧化铝、氧化钛等)、磷酸盐(ZrP2O7、Zr2O(PO4)2和Th4(PO4)4P2O7)、天然黏土矿物(膨润土和凹凸棒石黏土)和石灰性土壤等上的吸附。吸附质主要涉及Cs(Ⅰ)、Ni(Ⅱ)、Eu(Ⅲ)、Th(Ⅳ)和U(Ⅵ)等。
1 固体表面材料的表征
1.1 固体材料的结构、拓扑性质表征
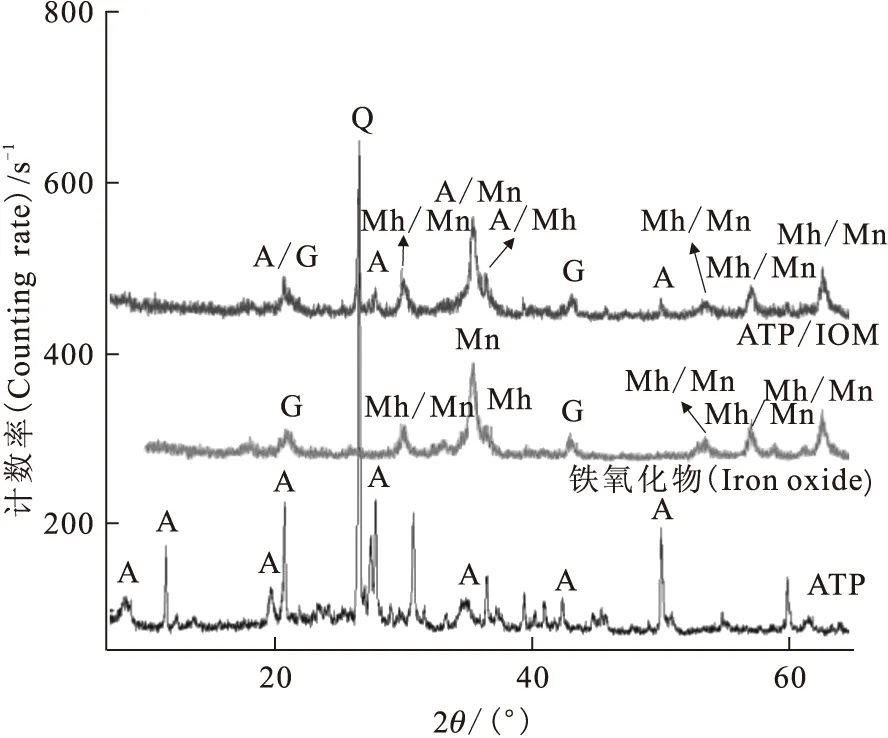
图1 凹凸棒石黏土和铁氧化物-凹凸棒石黏土磁性复合材料的XRD衍射图
为了研究吸附质与吸附剂的相互作用机理,需对吸附材料的结构、形貌、表面功能基团等信息进行详细的表征。通常XRD和SEM分别被用来表征吸附材料的基本结构和形貌。图1和图2分别为凹凸棒石黏土(ATP)和铁氧化物-凹凸棒石黏土磁性复合材料(ATP/IOM)的XRD衍射图和电子扫描电镜形貌图[3]。由图1可知,铁氧化物-凹凸棒石黏土磁性复合材料中除了2θ=20.7°、26.6°及42.5°处特征峰外,凹凸棒石其余特征峰均消失,说明凹凸棒石的结构可能因为铁氧化物的负载导致凹凸棒石晶格所谓的混乱[4]。在复合材料中其他峰位置与标准图谱JCPD中铁氧化物XRD谱非常接近:Fe3O4磁铁矿(89-3854,2θ=30.088°、35.439°、37.071°、43.07°、53.432°、56.958°和62.546°)或α-Fe2O3赤磁铁矿(89-5892,2θ=30.266°、35.651°、37.294°、43.332°、53.766°、57.319°和62.949°)[5]。另外,2θ=35.42°和36.62°处的峰位置可能是由ATP/IOM复合材料造成的。由图2可知,凹凸棒石呈纤维状且一些纤维聚集形成棒状,单个纤维直径约40~60 nm,长0.5~1 μm。而铁氧化物-凹凸棒石磁性黏土复合材料表面明显变得更紧凑、粗糙,粒径更小且边角较平滑,这可能是由于铁氧化物的引入使其形貌发生改变(图2(a)—(c))所致。

图2 凹凸棒石黏土和铁氧化物-凹凸棒石黏土磁性复合材料的SEM
图3为4种土壤样品的红外图谱对比,分别为除碳酸盐土壤(DCS)、Ca型石灰性土壤(CaCS)、吸附铀酰离子Ca型石灰性土壤(U(Ⅵ)-CaCS)和吸附铀酰离子除碳酸盐土壤(U(Ⅵ)-DCS)。由图3可知:3 620 cm-1和3 420 cm-1附近的峰分别对应AlO—H 和FeO—H的吸收峰;1 030 cm-1和467 cm-1处的峰为Si—O—Si振动,779 cm-1处的峰可能是由δ(O—Si)的伸缩振动引起;528 cm-1处的峰则对应O—P—O 的面外扭曲振动;而1 630 cm-1处的峰对应于水的弯曲振动[6-7]。对于Ca型石灰性土壤中在1 434 cm-1和875 cm-1处的吸收峰由CaCO3的官能团振动引起,但在除碳酸盐土壤中已经消失,表明碳酸盐组分被完全去除。CaCO3在1 434 cm-1和875 cm-1标准振动吸收在DCS、U(Ⅵ)-DCS和U(Ⅵ)-CaCS样品中消失,说明石灰性土壤中CaCO3组分参与U(Ⅵ)在其表面反应。

图3 4种土壤样品的红外图谱[8]
1.2 固体材料表面性质表征
1.2.1固体表面电荷产生 离子或有机质在固液界面的物理化学反应中,固体颗粒物的表面电荷和表面电势是决定吸附质在其表面上的吸附过程、微观吸附种态及吸附机理的重要参数。表面电荷/电势的改变对吸附质在固液界面上的化合态、表面反应活化能等均有重要的影响,从而导致离子或有机质在固液界面上的吸附反应方向和吸附/解吸反应动力学等的改变。固体颗粒物的表面主要通过以下3种方式形成表面电荷[9],即固体表面上的化学反应、固相表面的晶格缺陷和晶格内的同晶置换以及表面吸附疏水性的化合物或者表面活性剂。
(1)固体表面上的化学反应
固体表面一般都带有可离子化的官能团,如—OH、—NH2、—COOH、—OPO2H2、—SH等。可离子化官能团的质子转移作用可致使固体表面产生电荷,表观上表现出酸碱两性(即可发生质子化和去质子化反应)。由表面官能团上质子转移而产生的电荷,称之为可变电荷或者净质子电荷。可变电荷强烈依赖于体系pH值,在低pH值时固体表面主要以正电荷状态存在;当在较高的pH值条件下则固体表面主要表现出负电荷形态;而在某个特定的pH值下,固体表面电荷可以为零(此处对应的pH为零电荷点,pHpzc)。对于其他的固体表面可发生类似的质子转移反应,可写成普遍通式(1)和(2):
(1)

(2)
但当固体表面与存在于溶液中的溶质离子结合时亦可产生表面电荷。例如以下反应:

⟩Fe-OOPO3H-+H2O
(3)
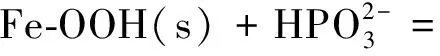
(4)
其中s表示固体[10]。
(2)固相的晶格缺陷和晶格内的同晶置换
固体晶格缺陷可使固体表面带电荷。当晶格内SiO2四面体中的四价Si被三价Al取代,或Al2O3中铝氧八面体中三价的Al被二价Mg、Fe等置换后,整个晶体带一个单位的负电荷,如图4所示。同晶置换作用是天然的黏土矿物骨架带有电荷及拥有一定离子交换容量的主要原因。由结构缺陷或者同晶置换作用而产生的表面电荷几乎不受固体所处溶液pH的影响,因此这类电荷被称之为结构电荷或者永久性电荷。

图4 三价Al对四价Si的同晶置换作用
(3)表面吸附疏水性的化合物或者表面活性剂
通过范德华力、氢键、色散力和其他化学物理作用使疏水性化合物或者表面活性剂吸附在固体表面上,也是导致固体表面带电荷的另一重要原因。最典型的例子就是氧化硅表面上吸附腐殖酸而致使氧化硅表面带有一定的电荷。图5表示不同固体表面电荷对其所处体系的pH值改变的趋势。结果表明,大多数固液界面都存在一定的表面电荷,且表面总电荷密度强烈依赖体系pH值。

图5 不同物质的表面电荷密度和电泳迁移率随pH值改变趋势[11]
1.2.2固体表面电荷密度和零电荷点 目前,连续电位滴定仍然是研究固体表面电位或电荷密度的重要方法之一。吸附剂表面位点密度、酸碱反应平衡常数以及零电荷点等参数都可以通过电位滴定获得。而研究吸附剂表面酸碱性质是定量描述放射性核素在其表面上吸附和解吸机理的必要前提。Guo等[12]利用连续电位滴定的方法详细探讨了Na基膨润土表面酸碱性质,发现同一离子强度下,样品的回滴曲线和滴定曲线之间存在明显的滴定滞后现象;而同一滴定方向(pH从4到9或pH从9到4)的滴定曲线间的差异则相对较小(图6)。ICP-OES测量发现,滴定过程中溶液中Al(Ⅲ)的最大浓度为2.5×10-5mol/L,说明矿物溶解作用对滴定结果的影响很小,且矿物溶解似乎也不是滴定滞后的主要原因。滴定滞后的最主要原因尚不清楚,仍有待进一步研究。Duc等[13]研究了样品的储存条件、CO2、固液比、滴加时间间隔、离子强度等因素对黏土矿物尤其是蒙脱石滴定的影响,发现采用湿法保存样品、惰性气体保护(消除CO2的影响)、缩短滴加时间间隔(约为10 min)等滴定策略可在一定程度上消除滴定滞后作用,提高测量的重现性和准确度。Na基膨润土表面过剩质子量(ΔQH)和Na基凹凸棒石表面电荷密度随pH变化示于图7。由图7(a)可知,不同离子强度下的3条滴定曲线没有交点且几乎平行,零电荷点随离子强度的减小而增大,这与文献中Na基蒙脱石的连续滴定结果一致[13-14]。但Fan等[15]利用连续电位滴定计算Na型凹凸棒石黏土表面零电荷点时却发现3种离子强度下滴定曲线几乎交于一点(pH≈6.02)。目前,固体表面的零电位点的表征方法有很多,除连续电位滴定以外,还有固体添加法[16]、电泳迁移率法(electrophoretic mobility)[17]和质量滴定法等[18]。钱丽娟等[18]利用质量滴定的方法求得3种磷酸盐的零电荷点(图8)。随着固液比逐渐增大,pH逐渐向一个定值接近。pH变化方向与起始的pH有关,最后趋向于一个交点,该点即为固体材料的pHpzc点。由质量滴定可知,ZrP2O7、Zr2O(PO4)2和Th4(PO4)4P2O7三种磷酸盐的pHpzc分别为3.5、4.2和6.8。

图6 金川Na基膨润土在0.1(a)、0.01(b)mol/L NaCl溶液中的酸碱滴定曲线
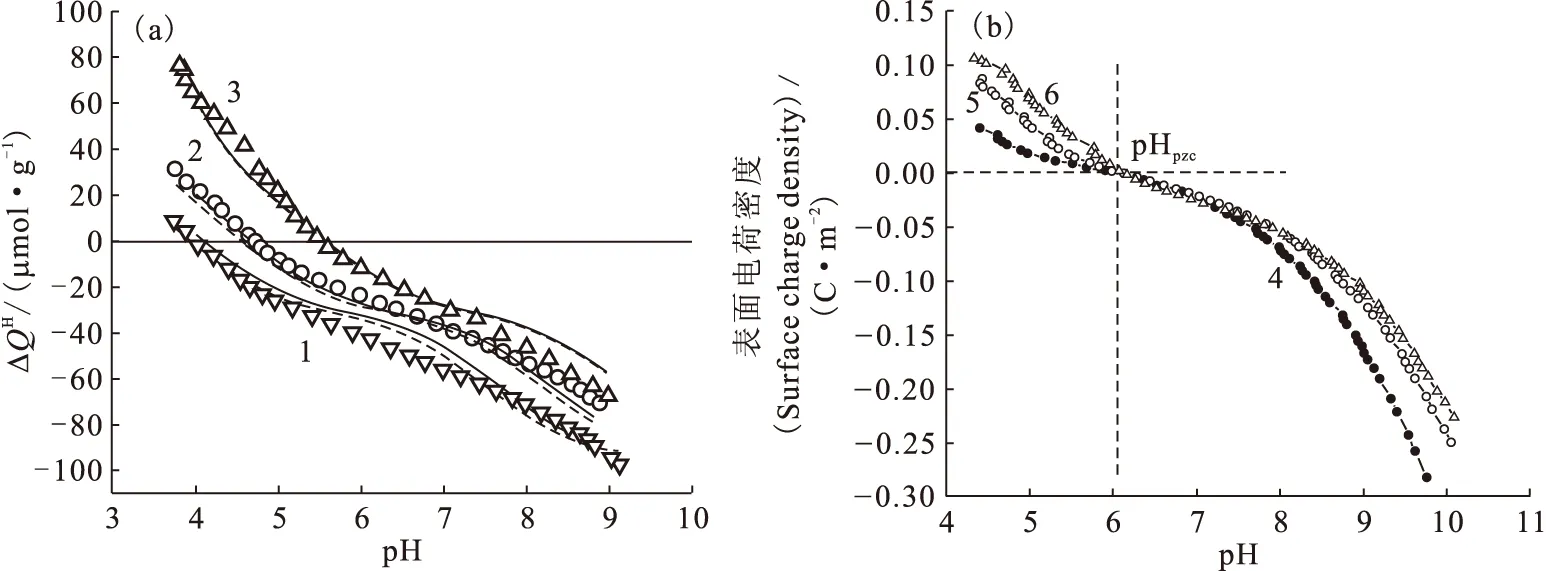
图7 Na基膨润土表面过剩质子量(a)[12]和Na基凹凸棒石表面电荷密度(b)[15]与pH关系图

图8 ZrP2O7(a)、Zr2O(PO4)2(b)、Th4(PO4)4P2O7 (c)的质量滴定曲线[18]
1.2.3表面配位模型 1998年Kraepiel等[19]提出两种表面配位模型以解释黏土矿物在不同离子强度下的平行滴定曲线。将黏土矿物看作一种不能穿透的固体物质,假设存在两种表面吸附位点:⟩X-和⟩S—OH,其中⟩X-位点所带的电荷与pH无关(永久性电荷),⟩S—OH位点所带电荷来自于表面质子化和去质子化反应,矿物的表面电荷密度为永久性电荷和可变电荷之和。矿物表面电荷和表面电势的关系可用Gouy-Chapman 理论计算[20]。假设:(1)蒙脱石表面除了层间吸附位点⟩X-(永久负电荷吸附位点),还存在两类边缘吸附位点,⟩Al—OH和⟩Si—OH,且N(⟩SiOH)/N(⟩AlOH)=2.0;(2)⟩Al—OH发生质子化和去质子化反应,而⟩Si—OH只发生去质子化反应[13];(3)Na基膨润土层间位点⟩X-完全被Na+占据,⟩X-位点的容量由测得的阳离子交换容量(CEC)值来确定。金川Na基膨润土表面发生的表面反应可用下列方程式表达:

(5)
(6)
(7)
(8)
(9)
依据上述表面反应,Na基膨润土表面质子过剩ΔQH可以表示为:

c(⟩XH)-c(⟩AlO-)-c(⟩SiO-))
(10)
其表面电荷ΔQcharge(mol/g)可以表示为:

c(⟩AlO-)-c(⟩SiO-)-c(⟩X-))
(11)
其表面电荷密度σ(C/m2)为:
(12)
式中:F是法拉第常数,96 485 C/mol;s是Na基膨润土的比表面积,m2/g;对于浸于对称电解质溶液的膨润土,其表面电势(ψ,V)计算公式为:
σ=(8RTεε0ce×103)1/2sin(ZeψF/2RT)
(13)
式中:ε是水的介电常数(25 ℃为78.5 C/(V·m));ε0为真空介电常数,8.854×10-12C/(V·m);ce是背景电解质浓度;Ze是背景电解质的化合价。反应 (5)~(9)的本征平衡常数可表示为:
(14)
(15)
(16)
(17)
(18)
假设表面物种的活度系数为1,溶液中物种的活度系数用Davies 方程计算,拟合参数如下:⟩X-、⟩Al—OH(s)、⟩Al—OH(w)和⟩Si—OH的位点密度分别为1.16×10-5(623 meq/kg)、1.88×10-8、5.69×10-7、1.18×10-6mol/m2,比表面积为53.6 m2/g。其他列入表1。

表1 FITEQL软件拟合滴定曲线结果
2 影响放射性核素在固液界面吸附的因素
影响金属离子或有机质在固液界面吸附行为的主要因素包括:(1)吸附剂固体本身物理和化学性质,如等电点、颗粒尺寸、固体的孔径大小、比表面积、表面官能基团等;(2)环境体系pH、离子强度、温度、压力、有机质、空气中各种气体的含量(如CO2)等对吸附质在环境介质中的物种分布、价态等都有较强的影响,另外这些因素同样对固体表面电荷和电位有着至关重要的影响;(3)吸附质的存在形态、价态、与溶液中各组分的相互作用等(包括与阴离子的配合作用和阳离子的竞争作用等)均对放射性核素在吸附剂表面上的吸附产生重要的影响。
2.1 pH和离子强度的影响

2.2 温度影响
温度是影响吸附质在固液界面上吸附行为的重要参数之一。通常温度对放射性核素的吸附有正影响,即吸附体系温度升高有利于放射性核素的吸附反应发生;但少数吸附质在固液界面上吸附与温度却成负相关,如Cs(Ⅰ)在天然高岭土、斜发沸石和花岗岩上的吸附。温度对吸附质在固液界面上的吸附影响可总结出以下经验:(1)温度升高使得吸附活化能增大,吸附熵增大,从而增大吸附质的最大吸附量;(2)吸附质在固液界面上的吸附过程可为放热或吸热过程,因此温度升高时反应产物的量可能增加也可能减少;但值得注意的是物理吸附总是伴随能量的释放,即温度升高总是削弱物理吸附反应;(3)当吸附质在固液界面上形成羟基配合物时,使得在不同pH下温度对吸附的影响不同。

图9 pH值对U(Ⅵ)在SiO2上的吸附影响(a)[21]和对Th(Ⅳ)在凹凸棒石上的吸附影响(b)[23]
2.3 腐殖质的影响
腐殖质是动植物经过长期的物理、化学、生物作用而形成的复杂天然有机物。天然水体系和土壤等都含有一定的腐殖质。腐殖质是大分子聚合物,化学结构复杂,其相对分子质量从几百到几千不等。Schnitzer和Schuppli[24-25]利用红外光谱系统研究了土壤腐殖质的主要官能团组成,发现主要构成官能团有羧基、醇羟基、酚羟基、氨基、醌型羰基和酮型羰基等。不同提取剂提取的腐殖质在相对分子质量、芳化度和主要官能团方面均存在一些差异。各种官能团的含量在不同来源的腐殖酸中差异很大。就胡敏酸和富啡酸比较来看,富啡酸含羰基、醇羟基、酚羟基和酮羰基的量较胡敏酸多,而胡敏酸含醌羰基的量要比富啡酸高。一般情况下,在低pH值下,腐殖酸对放射性核素在固液界面上吸附起促进作用;而在高pH值条件下,则可能会抑制放射性核素的吸附。反之,由于腐殖酸没有固定分子式和相对分子质量,其大分子结构形貌受到放射性核素、pH值、胶体颗粒等因素的影响,腐殖酸对放射性核素在氧化物-腐殖酸表面的作用机理影响不是非常清楚,还有待进一步研究。
3 微观光谱技术在放射性核素吸附研究中的应用
3.1 XAFS技术应用
长期以来,由于实验技术的局限性,放射性核素在固液界面体系中的化学行为研究多集中于宏观热力学和动力学层面上。20世纪80年代发展起来的同步辐射光源技术,使XAFS(X-ray absorption fine structure,X射线吸收精细结构谱)可以通过对固液界面体系中目标元素的原子周围环境进行分子水平上的研究,而获得无序表面组成和微观结构信息,为从分子水平上研究放射性核素在固液界面上的吸附反应、揭示界面反应机理等提供了重要的技术支持。XAFS具有下列无可比拟的优势:(1)EXAFS(扩展X射线吸收精细结构谱)现象来源于吸收原子周围最近邻的几个配位壳层作用,决定于短程有序作用,且可用于非晶态物质的研究,得到吸收原子近邻配位原子的种类、距离、配位数及无序度因子;(2)X射线吸收边具有原子特征,可对不同元素的原子周围环境分别进行研究;(3)利用强X射线或荧光探测技术可以测量到mg/kg浓度级别的环境样品;(4)EXAFS可用于固体、液体、气体等样品,一般不需要高真空,且不损坏样品。Fan等[15]利用EXAFS吸收谱证明了HA对Eu(Ⅲ)在凹凸棒石表面上吸附机理影响机制,发现不同HA的添加次序与Eu(Ⅲ)在固液界面上的微观结构紧密相关。不同HA添加次序下,Eu(Ⅲ)在凹凸棒石黏土表面的径向结构分布和k2χ(k)函数示于图10。通过对第一配位壳层(Eu-O)进行拟合,得到相关的结构参数列于表2。由表2可知,对于batch 1 (ATP+HA-Eu(Ⅲ))样品,原子间距d(Eu-O)≈0.241 1 nm(N=11.91,σ2=0.016 3);bacth 2(Eu(Ⅲ)+HA-ATP)样品的原子间距d(Eu-O)≈0.239 9 nm(N=11.66,σ2= 0.016 3);而batch 3 (ATP+Eu(Ⅲ)-HA)样品的d=0.232 1 nm(N=8.866,σ2=0.005 3)。值得注意的是batch 3样品结构参数与二元体系(Eu(Ⅲ)+ATP)非常相似(d(Eu-O)=0.231 4 nm,N=8.24,σ2=0.001 6);而batch 1和2的结构参数相似。此结果很好的说明了有机质的存在、体系pH值等因素对放射性核素在固液界面上的吸附机理有至关重要的影响。
3.2 XPS技术应用
单纯从静态批式实验数据和假设模型很难获得较准确的放射性核素在固液界面上的吸附物种和机理。XPS技术对固体表面元素分析具有高灵敏性等特点,因此,XPS技术可以很好的应用到放射性核素在固液界面上的吸附机理研究中,将XPS光谱、静态宏观实验和表面配位模型等相结合,必将促进人们对放射性核素在固液界面上微观吸附机理的进一步认识和理解。图11为pH分别为4.5和6.0时,吸附在凹凸棒石黏土和ATP/IOM复合材料上的U(Ⅵ)的U4f谱图。由图11可知:pH=4.5时,在U(Ⅵ)-凹凸棒石黏土二元体系中含378.57 eV和382.02 eV处的两种物种;而在U(Ⅵ)-ATP/IOM复合材料体系中则只有382.56 eV处的一种物种形成;当pH≈6.0时,在U(Ⅵ)-ATP/IOM复合材料体系中也存在两种物种(378.35 eV和382.05 eV处)。这一结果为后续的模型拟合奠定了基础。
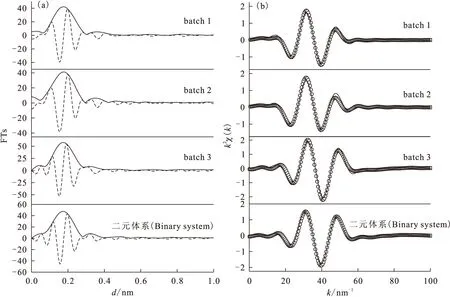
图10 不同HA添加次序下Eu LⅢ吸收边的径向结构函数(a)和k2χ(k)函数(b)[15]

表2 不同HA添加次序的EXAFS拟合结果
注(Notes):t=(20±1)℃,c(NaClO4)=0.01 mol/L;d,原子间距离(Interatomic distance);N,配位数(Number of neighbor oxygen atoms);σ2,Debye-Waller因子(Factor);ΔE0,能量位移(Energy shift);Rf,残留因子(Residual factor),Rf=∑k(k2xexp-k2xcalc)/∑k(k2xexp)
3.3 其他光谱技术应用
除了上述XAFS和XPS等光谱技术可以对放射性核素在固液界面上的微观结构和吸附机理进行原子或分子水平上的分析外,X射线能量色散谱(EDS)、时间分辨激光荧光光谱(TRLFS)、激光诱导荧光法(LIF)、红外(FTIR)、T-FTIR(透射)、漫反射(DR-FTIR)和衰减反射(ATR-FTIR)等光谱在固液界面上吸附的微观机理和结构研究方面同样有着重要的应用。SEM-EDS面分布是一种表面灵敏和直观的分析方法,能够直接得到目标元素在表面上的分布情况和在表面上的含量等信息。图12为不同背景电解质条件下吸附样品对Cs(I)吸附后的EDS分析。由图12可知:Cs主要是均匀分布在土壤颗粒的基面(basal plane)和边位点(frayed edge site,FES),说明Cs在土壤表面没有特定吸附区域。这种现象表明Cs(Ⅰ)在石灰性土壤上的吸附主要依靠静电作用的离子交换和范德华力。Cs的百分比随背景电解质阳离子的改变而发生规律性变换,阳离子对Cs的竞争能力遵从Mg2+>Ca2+≈ Na+的顺序。

图11 U(Ⅵ)在凹凸棒石黏土和ATP/IOM复合材料上的XPS U4f光谱[3]
图13为Eu(Ⅲ)在三种溶液中的荧光强度衰减曲线。在氧化铝存在下,半衰期110 μs 表明Eu(Ⅲ)的第一配位壳层含9个水分子,此结果与Eu3+离子在溶液中周围有9个水分子完全吻合,结果表明,在氧化铝溶液中Eu(Ⅲ)主要以水合Eu3+离子存在。210 μs表示Eu(Ⅲ)的第一配位壳层含4个水分子,说明氧化铝存在下,除Eu(Ⅲ)以水合离子存在于溶液中之外,还有部分Eu(Ⅲ)在失去5个水分子后被吸附到氧化铝颗粒表面上。在胡敏酸存在下,Eu(Ⅲ)只有一个荧光衰减时间,说明Eu(Ⅲ)与胡敏酸形成了一种稳定的配合物。在胡敏酸与氧化铝共同存在下,Eu(Ⅲ)则有两个不同的衰减时间,表明此时Eu(Ⅲ)形成了两种不同形态的配合物,Eu(Ⅲ)的第一配位壳层含5个水分子,说明Eu(Ⅲ)吸附在氧化铝的表层,在实验条件(如pH值、离子强度等)改变时有可能从氧化铝表层被解吸下来;而Eu(Ⅲ)第一配位壳层有2个水分子,说明Eu(Ⅲ)吸附在氧化铝的内层,即使实验条件改变也很难从氧化铝上被解吸下来,此吸附属于不可逆吸附。

图12 不同条件下Cs在石灰性土壤表面分布
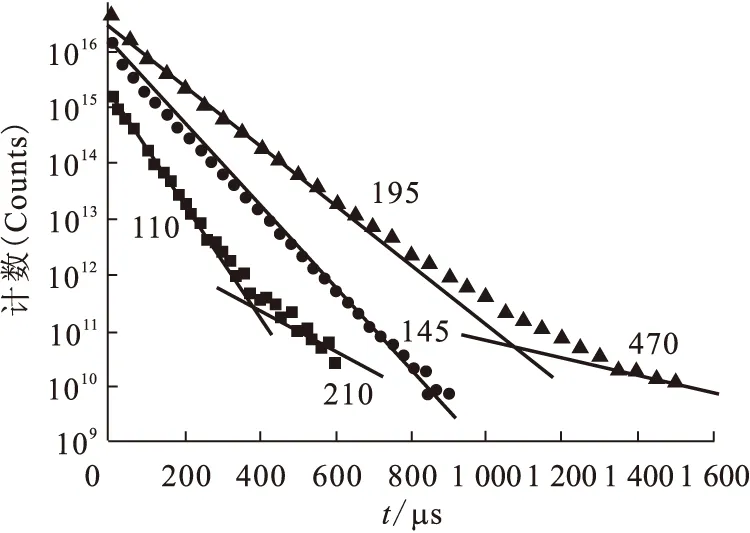
图13 Eu(Ⅲ)在三种溶液中的荧光强度随时间的变化关系[26]
4 放射性核素在固液界面的吸附机理


图14 Eu(Ⅲ)在凹凸棒石表面上的吸附种态随pH值的变化

表3 Eu(Ⅲ)在凹凸棒石表面上的吸附模型
5 结 论
本文主要以放射性核素在氧化物、磷酸盐和黏土表面上的吸附研究为例,综述了放射性核素在固体吸附剂表面上的宏观吸附实验和微观表征方法。影响放射性核素在固液界面上吸附的因素多且复杂,如pH值、离子强度、温度和有机质等。因此选择合适、正确的吸附模型对放射性核素在固液界面上的吸附研究具有重要的意义。表面配位模型在放射性核素吸附研究领域中取得了很大的成功,这主要是因为其模型能方便、合理地描述和解释实验数据。不同吸附材料由于其自身的结构、形貌和电荷等性质的差异,在微观吸附种态和吸附机理的探讨和论证中,还必须采用先进的光谱技术对微观吸附过程进行分子水平上的研究,如EXAFS、XPS、TRLFS和EDS等对放射性核素在氧化物、磷酸盐和黏土表面上吸附机理的探讨。然而,这些分析技术在环境样品的研究中仍然受到诸多因素的限制,仍需要探索和发展新的研究方法和手段,为得到正确和真实的吸附机理研究提供直接的、关键性的证据。
[1]Wang Y Q,Fan Q H,Li P,et al.The Sorption of Eu(Ⅲ)on Calareous Soil:Effects of pH,Ionic Strength,Temperature,Foreign Ions and Humic Acid[J].J Radioanal Nucl Ch,2011,287:231-237.
[2]Fan Q,Shao D,Lu Y,et al.Effect of pH,Ionic Strength,Temperature and Humic Substances on the Sorption of Ni(II)to Na-Attapulgite[J].Chem Eng J,2009,150(1):188-195.
[3]Fan Q H,Li P,Chen Y F,et al.Preparation of Attapulgite/Iron Oxide Magnetic Composites and Application in Removal U(Ⅵ)From Aqueous Solution[J].J Hazard Mat,2011,192(3):1 851-1 859.
[4]Zhao G X,Zhang H X,Fan Q H,et al.Sorption of Copper(Ⅱ)Onto Super-Adsorbent of Bentonite-Polyacrylamide Composites[J].J Hazard Mat,2010,173(1-3):661-668.
[5]Chen C L,Wang X K,Nagatsu M.Europium Adsorption on Multiwall Carbon Nanotube/Iron Oxide Magnetic Composite in the Presence of Polyacrylic Acid[J].Environ Sci Technol,2009,43:2 362-2 367.
[6]Oonk S,Slomp C P,Huisman D J,et al.Geochemical and Mineralogical Investigation of Domestic Archaeological Soil Features at the Tiel-Passewaaij Site,the Netherlands[J].J Geochem Explor,2009,101:155-165.
[7]Fan Q,Li Z,Zhao H,et al.Adsorption of Pb(Ⅱ)on Palygorskite From Aqueous Solution:Effects of pH,Ionic Strength and Temperature[J].Appl Clay Sci,2009,45(3):111-116.
[8]Zhang Y Y,Zhao H G,Fan Q H,et al.Sorption of U(Ⅵ)Onto a Decarbonated Calcareous Soil[J].J Radioanal Nucl Ch,2011,288(2):395-404.
[9]Anderson M A,Rubin A J.Adsorption of Inorganic at Solid-Liquid Interface[M].Ann Arbor,Michigan:Ann Arsor Sci,1981,Charp.1.
[10]陶祖贻,杜金洲.氧化物/水界面上的表面络合模型[J].离子交换与吸附,1994,10(2):112-118.
[11]Stumm W.Chemistry of the Solid-Water Interface:Processes at the Mineral-Water Interface in Natural Systems[M].New York:Wiley-Interscience,1992:43.
[12]Guo Z J,Xu J,Shi K L,et al.Eu(Ⅲ)Adsorption/Desorption on Na-Bentonite:Experimental and Modeling Studies[J].Colloid Surf A,2009,339(1-3):126-133.
[13]Duc M,Gaboriaud F,Thomas F.Sensitivity of the Acid-Base Properties of Clays to the Methods of Preparation and Measurement:2.Evidence From Continuous Potentiometric Titrations[J].J Colloid Interf Sci,2005,289(1):148-156.
[14]Tertre E,Berger G,Simoni E,et al.Europium Retention Onto Clay Minerals From 25 to 150 ℃:Experimental Measurements,Spectroscopic Features and Sorption Modelling[J].Geochim Cosmochim Acta,2006,70(18):4 563-4 578.
[15]Fan Q H,Tan X L,Li J X,et al.Sorption of Eu(Ⅲ)on Attapulgite Studied by Batch,XPS,and EXAFS Techniques[J].Environ Sci Technol,2009,43(15):5 776-5 782.
[16]Srivastava V C,Mall I D,Mishra I M.Adsorption of Toxic Metal Ions Onto Activated Carbon Study of Sorption Behaviour Through Characterization and Kinetics[J].Chem Engin Process,Process Intensification,2008,47:1 269-1 280.
[17]Drot R,Lindecker C,Fourest B,et al.Surface Characterization of Zirconium and Thorium Phosphate Compounds[J].New J Chem,1998,22:1 105-1 109.
[18]钱丽娟,胡佩卓,蒋正江,等.pH、富里酸和温度对铀酰在ZrP2O7上的吸附影响[J].中国科学 化学,2010,40:1 712-1 720.
[19]Kraepiel A M L.On the Acid-Base Chemistry of Permanently Charged Minerals[J].Environ Sci Technol,1998,32:2 829-2 838.
[20]Hoch M,Weerasooriya R.New Model Calculations of pH-Depending Tributyltin Adsorption Onto Montmorillonite Surface and Montmorillonite-Rich Sediment[J].Environ Sci Technol,2004,39(3):844-849.
[21]Zhang H,Wen C,Tao Z,et al.Effects of Nitrate,Fulvate,Phosphate,Phthalate,Salicylate and Catechol on the Sorption of Uranyl Onto SiO2:A Comparative Study[J].J Radioanal Nucl Ch,2011,287:13-20.
[22]许君政,范桥辉,白洪彬,等.离子强度、温度、pH和腐殖酸浓度对Th(Ⅳ)在凹凸棒石上吸附的影响[J].核化学与放射化学,2009,31(3):179-185.
[23]Wu W S,Fan Q H,Xu J Z,et al.Sorption-Desorption of Th(Ⅳ)on Attapulgite:Effects of pH,Ionic Strength and Temperature[J].Appl Radiat Isot,2007,65(10):1 108-1 114.
[24]Schnitzer M,Khan S U.Humic Substances:Chemistry and Reactions In:Soil Organic Matter[M].Amsterdam:Elsevier,1978:1-64.
[25]Schnitzer M,Schuppli P.Method for the Sequential Extraction of Organic Matter From Soils and Soil Fractions[J].Soil Sci Soc Amer J,1989,53(5):1 418-1 424.
[26]王祥科,郑善良.荧光衰减光谱法研究Eu(Ⅲ)在氧化铝表面的化学形态[J].核化学与放射化学,2005,27(2):108-112.
[27]Naveau A,Monteil-Rivera F,Dumonceau J,et al.Sorption of Europium on a Goethite Surface:Influence of Background Electrolyte[J].J Contam Hydrol,2005,77(1-2):1-16.
[28]Bradbury M H,Baeyens B.Sorption of Eu on Na- and Ca-Montmorillonites:Experimental Investigations and Modelling With Cation Exchange and Surface Complexation[J].Geochim Cosmochim Acta,2002,66(13):2 325-2 334.
