聚亚烷基二醇制备方法概述
2012-01-04程亮
程亮
(中国石油大连润滑油研究开发中心,辽宁大连 116032)
聚亚烷基二醇制备方法概述
程亮
(中国石油大连润滑油研究开发中心,辽宁大连 116032)
介绍了聚亚烷基二醇(PAG)的主要特性和在润滑油中的应用情况。对PAG的发展历史、制备方法及其反应机理研究现状进行了简要总结。重点介绍了目前制备PAG的几种聚合方法,包括阳离子聚合、阴离子聚合、配位聚合,并对每种聚合方法的机理、特点及催化剂的研究现状进行了总结。
聚亚烷基二醇;润滑;阳离子聚合;阴离子聚合;配位聚合
0 引言
聚亚烷基二醇(Polyalkylene glycols(PAG),也称做聚醚)是由环氧化合物及其衍生物共聚或均聚而成的主链中含有醚键结构的高分子聚合物,其主要结构单元由环氧乙烷(EO)、环氧丙烷(PO)、环氧丁烷(BO)或四氢呋喃(THF)组成。聚亚烷基二醇共聚物根据单体序列的分布可以分为无规聚合物和嵌段聚合物,其中由于无规聚醚具有优异的高低温性能、氧化稳定性、黏温性、清净性、剪切稳定性、低的倾点及化学性质稳定等优点而被成功地应用在润滑领域,如表1所示。

表1 PAG在润滑领域的应用及其特性

续表
对于聚醚的合成一直是人们研究的热点。早在1859年,Lourenco等人通过封管技术,将乙二醇与1,2-二溴乙烷的混合物直接加热至115~120℃,首次获得了具有醚键结构的低分子量聚合物。在同一年,Wurtz等人分离得到了环氧乙烷和水的聚合产物—聚乙二醇。1863年,Wurtz等人首次报道了室温下少量的碱或氯化锌可以加速环氧乙烷的开环聚合。直到1927年,Levene和Walti等人才首次报道通过加热到165℃可以实现环氧丙烷的聚合。1929年,Staudinger和Schweitzer报道了钾、四氯化锡、三甲胺等可以在温和的条件下促进环氧乙烷的开环聚合,并且对反应的速率进行了研究,同时指出四氯化锡也可以实现环氧丙烷的开环聚合,此外Staudinger通过与Svedberg合作,首次得到了不同分子量的聚合产物。紧接着在I.G.Farbenindustrie的Indigo实验室研究人员发现金属铁或含铁的化合物可以高效促进环氧乙烷的开环聚合。在随后几十年的发展中,各种催化剂不断涌现,得到了分子量高、分布窄和不饱和度低的聚醚类产品。目前主要存在三种催化环氧烷烃开环聚合的方法,分别是:酸催化聚合、碱催化聚合和配位催化聚合。本文分别予以评述。
1 酸催化聚合(阳离子聚合)
早在1939年,Meerwein等人[1]就完成了利用Lewis酸催化环氧烷烃生成二聚或三聚的小分子聚合物的研究,但是人们对Lewis酸是否可以催化环氧烷烃聚合仍然没有统一的认识。直到20世纪五六十年代,Dainton[2]、Plesch[3]、Kennedy[4]等人相继发表了Lewis酸对环氧烷烃的催化聚合的工作,人们对酸催化环氧烷烃得到高分子聚合物有了统一的认识。酸催化聚合的引发剂[2-3]通常为Brønsted酸或Lewis酸(在使用Lewis酸作为引发剂时,需要加入少量烷基卤化合物或含活泼氢的化合物作为共引发剂)[5]。聚合过程分为四个阶段,分别是起始剂形成、链引发、链增长和链终止,如图1所示。在起始剂形成阶段,Brønsted酸或Lewis酸与共引发剂形成起始剂。在链引发阶段,环氧化合物与引发物种形成氧鎓离子,在醇或烷基卤的作用下进行SN2开环反应实现C-O键断裂,形成链增长的活性物种。接着它与反应单体进行链增长,形成长链聚合物。在链终止阶段,反应体系可以通过两种方式终止反应:第一种是阴离子(或卤素)转移。即活性体中的阴离子(或卤素)向单体转移;第二种是氢离子转移。但同时可产生不饱和大分子产物即氢离子的重排导致活性链失活,从而终止反应。在酸催化的聚合反应中,反应的速率和聚合产物的平均分子量主要由起始剂中离子对的结构、阴离子的碱性、反应温度和溶剂决定的。需要指出的是,在其他条件相同的情况下,聚合链增长和链终止的相对速率决定了聚合物的平均分子量大小。

图1 酸催化聚合示意
酸催化聚合存在反应快、分子量低及分子量分布宽等缺点。目前在四氢呋喃的开环聚合反应中应用较多。
2 碱催化聚合(阴离子聚合)
碱催化聚合是目前工业上普遍采用的方法,属于典型的SN2开环反应,常用引发剂[6]一般是碱金属或碱土金属的氢氧化物、碱金属醇盐,如甲醇钠或甲醇钾等,引发方式如图2所示。当单独使用醇钠或醇钾作为引发剂时,聚合反应的速率非常慢,当加入烷基醇作为共引发剂时,聚合反应的速率则大幅度提高,同时聚合物的分子量分布变窄[7]。这主要是由于它们可以形成络合物,从而弱化了醇盐离子对之间的作用力(图3(1)),更有利于与环氧烷烃形成有效的三元络合物(图3(2))。

图2 碱催化聚合引发
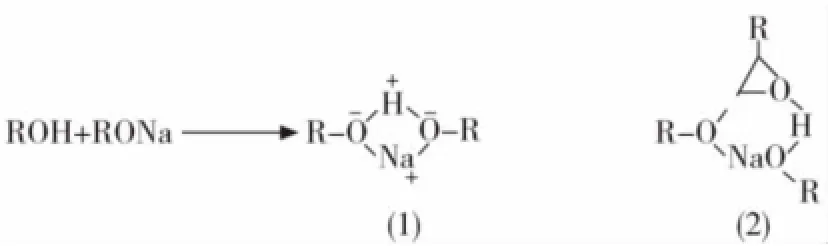
图3 醇盐形成示意
表2列出了环氧乙烷和环氧丙烷与各种醇的加成反应速率,可以看出:(1)一级醇的反应活性大于二级醇;(2)环氧乙烷的加成速率大于环氧丙烷; (3)β-烷氧基乙醇的加成速率不受烷氧基上取代基团的影响。
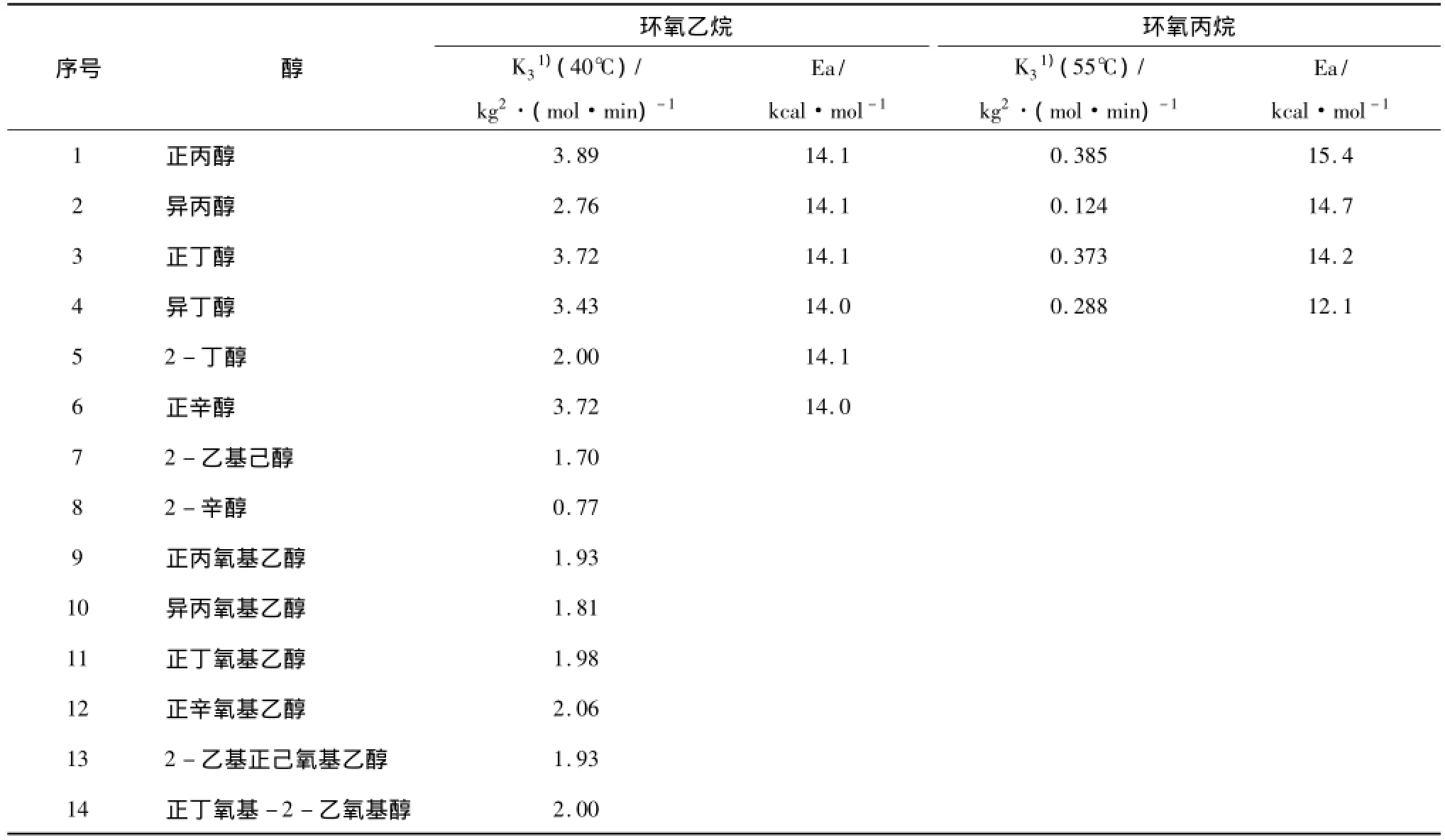
表2 环氧乙烷和环氧丙烷与各种醇的加成速率
碱催化聚合反应的链增长方式与酸催化聚合类似,然而终止反应一般不会自发进行,需要加入少量的水或者醇。环氧乙烷和环氧丙烷均聚产物的端基分别是一级羟基和二级羟基。在碱催化聚合的过程中,开环反应通常发生在含取代基较少的C-O键之间,断裂后的烷基氧原子仍然具有很高的活性,因此容易发生一些副反应。对于环氧乙烷来说,活性端基烷氧基可以夺取聚合物链上的氢原子,从而形成端基不饱和链和新的聚合物活性链(图4(1))。对于环氧丙烷来说,位于端基的烷基氧原子可以通过两种方式夺取聚合物链上得氢原子,从而得到烯丙基醚或者丙烯基醚(图4(2))。碱性离子可以引起环氧丙烷单体的重排而生成烯丙基醇或者生成烯丙基醚的活性聚合链(图4(3))[8]。因此,为了减少副反应的发生,首先必须保持比较低的反应温度(通常低于100℃);其次是保持在反应过程中一直有过量的单体存在。
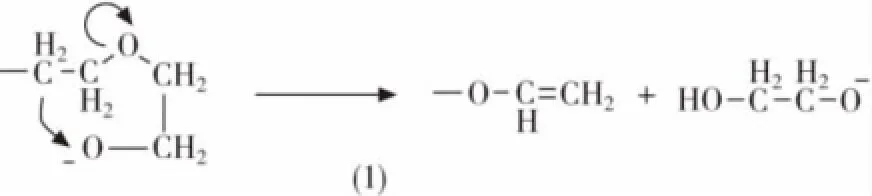

图4 碱催化聚合可能的副反应
碱催化聚合与酸催化聚合相比,具有反应速率慢(氯甲基环氧丙烷、羟甲基环氧丙烷除外[9-10])、分子量相对高和分子量分布较窄等特点。表3列出了酸催化聚合与碱催化聚合的反应速率比较(以高氯酸与乙醇钠为引发剂)。可以看出,在碱催化聚合中,当环氧烷烃上连接有吸电子基团时,反应具有较高的活性(表3)。随着环氧烷烃主链的增长,其反应活性逐渐降低。然而在酸催化聚合中则完全相反。这是由于两者的速率决定步不同(如图5)。在酸催化聚合中,质子可以与氧原子形成鎓盐,弱化了C-O键,从而有利于亲核试剂的进攻;而在碱催化聚合中,吸电子基团的存在使得亲核试剂很容易进攻碳正离子。

表3 不同环氧烷烃在酸和碱催化下的聚合速率比较
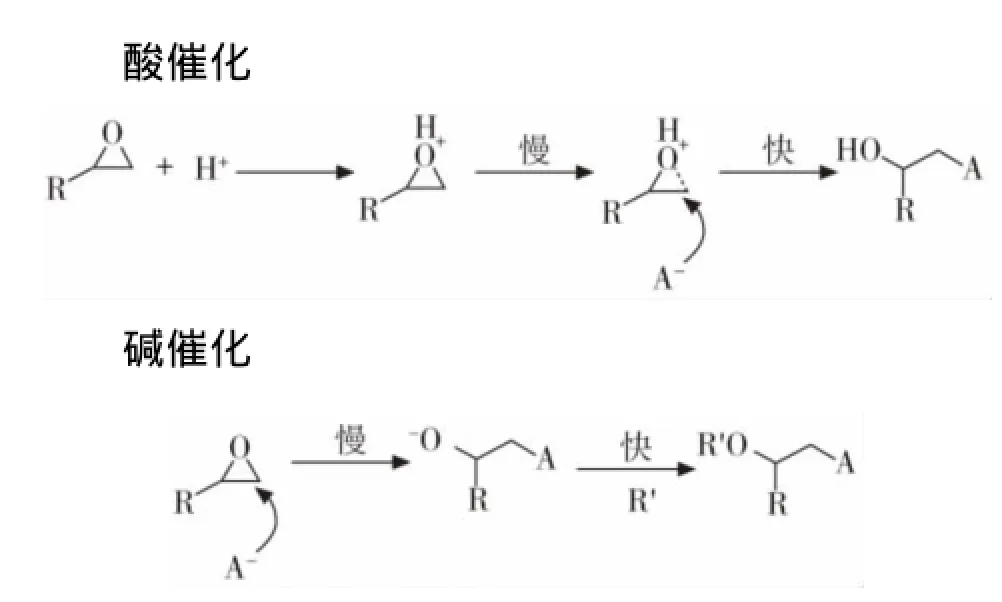
图5 酸、碱催化聚合速率决定步
3 配位催化聚合
配位催化聚合即金属络合聚合[7],金属(如Li、 Zn、Al、Sn、Fe、Mg、Ca等)与配体(如:OH、OR、X、OS-nR3、NH2等)形成络合物,络合物通过与环氧烷烃上的氧原子(或单体)络合活化环氧烷烃分子,配体亲核进攻已活化的环氧烷烃分子导致环氧烷烃开环,形成活性物种,继而进行链增长,形成聚合物长链,如图6所示。

图6 配位催化聚合示意
1952年,Pruitt和Baggett等人将三氯化铁和环氧丙烷组成的络合物,成功的用于环氧丙烷的聚合反应中[11]。这是首次明确了配位催化聚合的概念,并且使我们对这一催化体系有了更好的了解。随后,大量的金属催化剂不断涌现,根据Furukawa和Saegusa的研究[12],这些催化剂可以分为两类:第一类是通过形成Metal-O配位键,活化C-O键,从而实现环氧烷烃的开环,主要包括Al(OR)3,Zn (OR)2,Ca0.5[Al(OR)4],Al(OR)3·ZnCl2,Al (OR)3·H2O,FeCl3/PO,MgR2,RMgX,ZnR2· CaF2,Al2O3·ZnR2,ZnEt2·MgO等;第二类是通过金属盐(一般是碱土金属)与配体(一般为少量的水或者硝酸根、硫酸根等强酸性阴离子)组成的络合物与环氧烷烃分子络合,从而实现环氧烷烃的开环,这类金属催化剂主要包括CaO,SrCO3,Sr(OR)2,Ca (OR)NH2,Ca(NH2)2,Ca(NH3)6等,这类催化剂需要少量环氧烷烃化合物来引发。一般来说,第一类金属催化剂对于环氧丙烷和环氧丁烷的开环聚合具有很好的催化效率;然而,第二类催化剂对于环氧乙烷的开环聚合具有很高的反应速率。经过几十年的发展金属催化剂总结起来可以归纳为以下几类:烷基金属催化剂、金属卟啉催化剂、双金属氰化物催化剂。
3.1 烷基金属类催化剂
烷基金属类催化剂是最早发展起来的金属催化剂,主要包括ZnEt2、EtMgBr、Et3Al等。在这一类催化剂发现初期,这类催化剂并没有表现出很高的催化活性,并且增大反应中催化剂的浓度时,催化活性反而降低。Furukawa等人研究发现,当向这类催化体系中加入少量的水、醇或氧气时,这类催化剂表现出很高的活性[13]。以ZnEt2/H2O体系为例,在这个体系中,ZnEt2与水反应生成无定形的烷氧基锌(图 7(1)),当催化环氧丙烷的开环聚合时,烷氧基锌与环氧丙烷形成四元环过渡态,活化C-O键,实现开环聚合(图7(2))。然而晶态烷氧基锌是没有催化活性的[14],原位生成的烷氧基锌可能以单体、二聚体、三聚体或多聚体的形式存在,单体、二聚体形式均可以与环氧烷烃形成环状过渡态,然而在三聚体或多聚体中,由于锌没有空余的配位点而不能与环氧烷烃形成环状过渡态,因而具有很低的催化活性(图8)。因此,当原位生成的烷基锌是以单体或二聚体的形式存在时,其具有很高的催化活性,随着催化剂放置时间的延长,单体或二聚体会向三聚体或多聚体方向转化,同时催化活性降低。
一般来讲,这类催化剂要具有高活性须满足以下两点,(1)催化剂中含有M(OR)n活性物种;(2)催化剂是无定形态的。
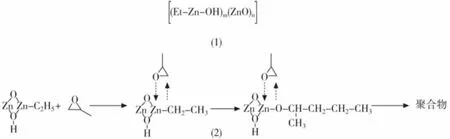
图7 ZnEt2/H2O体系催化PO开环聚合
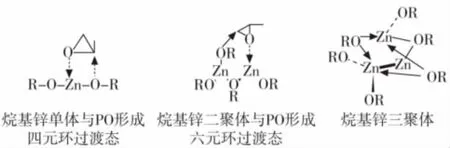
图8 烷氧基锌聚集态
3.2 金属卟啉催化剂
金属卟啉催化剂是70年代末发展起来的一类配位催化剂[15],它由于可以制备出分子量分布窄、可控分子量的环氧聚合物和副反应少而备受关注。常见的体系是M-卟啉-X(M为铝、锌、锰、钛、钴等金属;X为Cl、OH、OR、O2CR、SR、CH2CHO等)[16],结构如图9所示。这类催化剂引发聚合的机理一般是在金属原子与轴向配体之间插入环氧烷烃,形成卟啉醇化物活性物种来完成环氧烷烃的开环反应。当向反应体系中加入质子性化合物(如酸,醇,水等)时,反应的链增长不会被终止,而是生成另外一种新的引发剂,可以继续实现环氧烷烃的开环反应(图10)。因此这类催化剂引发的聚合反应也被称为永活性聚合[17]。
由于环氧烷烃的开环聚合反应发生在M-X键上,所以金属原子周围的电子密度决定着链引发与链增长的速率。以卟啉-铝催化剂催化β-羰基环氧丙烷的开环聚合为例,当催化剂中的R1=R2= OMe时,它的催化活性大于R1=R2=H时的活性;然而当R1=R2=Me时的催化活性也远远小于R1= R2=OMe时的活性[18],但是当R1=Ph,R2=H时此催化剂没有活性。因此电子效应与空间位阻效应共同决定了催化剂的活性。另外,卟啉的大环结构使得各中心金属离子彼此孤立,难于发生分子间相互作用,从而使与金属中心相结合的环氧烷烃基团表现出相同的反应活性,导致环氧烷烃在开环聚合过程中具有相同的链引发和链增长速率,能够合成出单分散的高聚物[19]。
这类催化剂的特点可以概括为:(1)Lewis酸的存在可以促进聚合反应的发生;(2)聚合过程不容易失活;(3)可以催化许多种类的环氧单体进行开环聚合。虽然金属卟啉催化剂在环氧烷烃的开环聚合反应中具有很多优势,但反应速度过慢,反应中催化剂不稳定等缺点限制了它的大规模使用。近年来,人们开始设计位阻型和高分子负载型卟啉催化剂,具有潜在的应用前景。

图9 金属卟啉催化剂结构
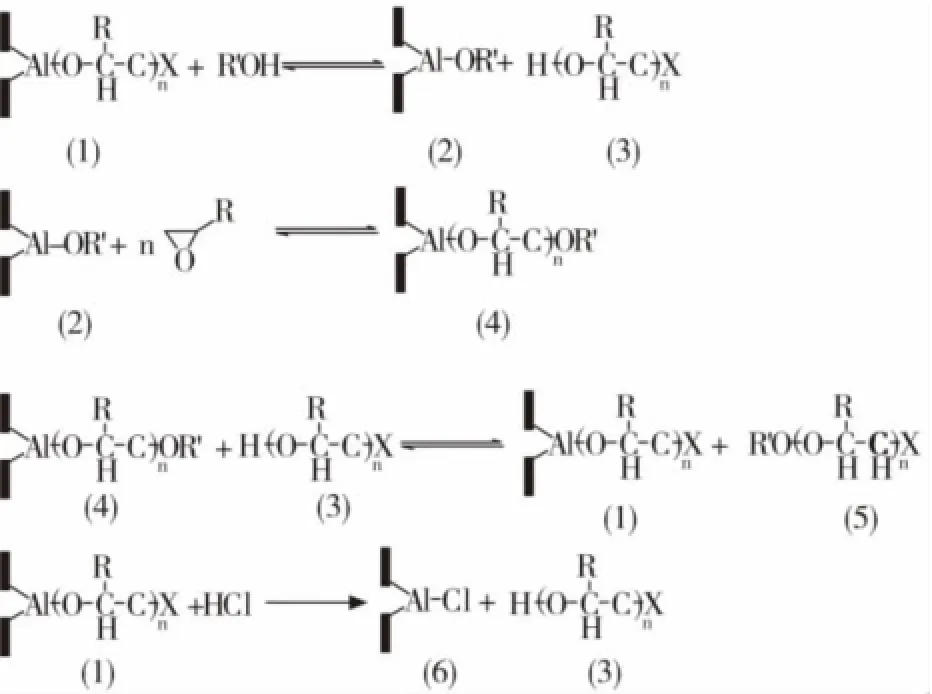
图10 金属卟啉催化剂催化环氧烷烃开环示意
3.3 双金属氰化物催化剂(DMC)
早在20世纪60年代,General Tire公司首先发现双金属氰化物催化剂可以用于环氧烷烃的开环聚合反应[20],但是并没有引起人们的注意。直到20世纪80年代中期,ARCO[21]、Shell[22]、Asahi Glass[23]等公司相继报道DMC催化剂,人们才开始真正关注它。
与其他的碱性催化剂相比,DMC催化剂可以获得低不饱和度、分子量分布窄、低黏度的聚醚产品。同时DMC催化剂对空气不敏感,催化活性高,对聚合条件要求较为宽松,具有较好的工业前景。
对于DMC的催化机理,目前还没有统一的定论。早在1966年,Jack等人[24]指出,以六氰铁酸钾、氯化锌和1,4-二氧六环为原料来制备DMC催化剂,在制备过程中Fe3+、Cl-和1,4-二氧六环之间存在氧化还原反应。Schuchardt[25]等人在DMC催化环氧丙烷的聚合机理的研究中指出,催化剂首先与环氧烷烃作用生成活性中心,紧接着起始剂与活性中心发生交换作用生成聚醚。Robert等人[26]认为首先催化剂形成以二价金属为主的活性中心,然后环氧烷烃与金属离子络合,开环插入形成MX-L,继而实现链增长。在链增长的过程中,环氧烷烃单体可以有一级插入和二级插入两种方式。一级插入使含取代较多的C-O键断裂,形成伯羟基。二级插入则相反,最后形成仲羟基。根据实验发现,对于环氧丙烷,一级与二级插入之比为1∶10,主要产生仲羟基。对于无取代基的环氧乙烷,则只能得到伯羟基。刘晓华等人[27]分别用乙醚、乙二醇二甲醚和二乙二醇二甲醚为有机配位体,以六氰钴钾和氯化锌为原料,制备了一系列DMC催化剂。结果发现金属离子与有机配体之间作用力的强弱对催化剂的活性有较大影响。同时利用XRD和X射线光电子能谱等分析手段研究了DMC催化剂的结构和表面价态。结果表明,在催化剂制备过程中,未发生氧化还原反应,有机配位体只是与Zn2+配位,这种配位破坏了Zn3[Co(CN)6]2·H2O晶体结构,从而形成无定形态固体。因此有机配体能否与Zn2+配位是决定催化剂活性的关键,因此得出结论:(1)DMC催化剂中的活性结构和与氧配位的Zn2+数目有关。氧与锌的配位数越高,催化剂的活性越好;(2)Co在DMC催化剂中起到了活化Zn-N键的作用。这种活化作用由钴离子对氰基的强相互作用引起;(3)在催化剂激活过程中存在氧原子与锌原子的加成反应,通过这种加成反应生成聚合反应真正的活性中心,该活性中心可能为5或6个氧原子配位的锌离子。
然而,黄亦军等人[28]则认为DMC催化过程属于弱阳离子配位机理。以Zn3[Co(CN)6]2为例,当使用Zn3[Co(CN)6]2作为催化剂,水作为起始剂,按催化剂/单体/起始剂(质量比)=1/2/0.05的比例进行投料,聚合反应后,所得混合物在去除单体后,红外检测发现没有新的M-O或M-C键生成,这表明没有发生配位-插入机理。
有机配体在DMC催化剂中的作用是不容忽视的,目前,人们通过改变有机配体从而获得活性不同的DMC催化剂。同时,当DMC催化剂络合一些低聚物时,也表现出令人满意的活性。
3.4 其他类催化剂
在研究环氧烷烃的开环聚合的反应中,也有另外一些催化剂涌现出来,其中包括:双金属烷氧基类催化剂[29-30]、稀土类催化剂[31-32]、磷腈类催化剂[33-34]、聚合有机金属氧化物类催化剂[35]等。
4 结束语
PAG由于含有C-O-C醚键而在润滑领域有很广泛的应用,因此人们对于它的制备方法相当关注。目前存在着三种主要制备方法:酸催化聚合、碱催化聚合、配位聚合。其中碱催化聚合是工业化最成熟、应用最多的方法,但是配位聚合是目前最先进的方法,也是人们重点关注的方法。酸催化聚合目前由于成本高、后处理复杂而被逐渐淘汰。
人们在研究配位聚合的过程中,对金属如何参与反应的机理不太清楚,只是停留在假设的层面上,因此需要在筛选催化剂的同时,研究其作用机理,这将有助于制备出高效的催化剂。
[1]Meerwein H,Battenberg E,Gold H,et al.Über Tertiäre Oxoniumsalze II[J].J Prakt Chem,1939(154):83-156.
[2]Dainton FS,Devlin TR E,Small PA.The Thermodynamics of Polymerization of Cyclic Compounds by Ring Opening[J].Trans Far Soc,1955(51):1710-1720.
[3]Plesch PH.The Chemistry of Cationic Polymerization[M].New York:Pergamon Press,1963.
[4]Kennedy J P,Marachal E.Carbocationic Polymerization[M].New York:Wiley&Sons,1968.
[5]Mijangos F,León L M.Ethylene Oxide Polymerization by Triphenylmethyl Tetrafluorborate in Nitrobenzene at 25°[J].European Polymer Journal,1983(19):29-32.
[6]Armengol E,Corma A,Fernández L,et al.Acid Zeolites as Catalysts in Organic Reactions.Acetylation of Cyclohexene and 1-Methylcyclohexene[J].Applied Catalysis A: General,1997(158):323-335.
[7]Wojtech V B.Zur Darstellung Hochmolekularcer Polyäthylenoxyde[J].Makromol Chem,1963(66):180-195.
[8]Gallet G,Carroccio S,Rizzarelli P,et al.Thermal Degradation of Poly(Ethylene Oxide-Propylene Oxide-Ethylene Oxede)Triblock Copolymer:Comparative Study by SEC/MALDI-TOF-MS and SPME/GC-MS[J].Polymer,2002(43):1081-1094.
[9]Tsarevsky N V,Bencherif SA,Matyjaszewski K.Graft Copolymers by a Combination of ATRPand Two Different Consecutive Click Reactions[J].Macromolecules,2007(4): 4439-4445.
[10]Zhang Z,Yin H,Fang X.Quantum Chemical Study on Anionic Polymerization Mechanism of Propylene Oxide[J].Journal of Zhejiang University-Science A,2006(7): 325-329.
[11]Pruitt M E,Baggett JM.Catalysts for the Polymerization of Olefin Oxides:US,2706181[P].1955.
[12]J Furukawa,T Saegusa.Polymerization of Aldehydes and Oxides[M].New York:Wiley-Interscience,1963.
[13]Furukawa J,Tsuruta T,Sakata R,et al.Polymerization of Propylene Oxide by Diethylzine in the Presence of Cocatalysts[J].Makromol Chemie,1959(32):90-94.
[14]Wegener G,BrandtM,Duda L,et al.Trends in Industrial Catalysis in the Polyurethane Industry[J].Applied Catalysis A:General,2001(221):303-335.
[15]Takeda N,lnoue S.Polymerization of 1,2-Epoxypropane and Copolymerization with Carbon Dioxide Catalyzed by Metalloporphyrins[J].Makromol Chem,1978(179):1377-1381.
[16]Schütz C,Dwars T,Schnorpfeil C,et al.Selective Polymerization of Propylene Oxide by a Tin Phosphate Coordination Polymer[J].Journal of Polymer Science Part A:Polymer Chemistry,2007(45):3032-3041.
[17]Odian G G.Principles of Polymerization[M].New York: Wiley&Sons,2004.
[18]Sugimoto H,Aida T,Inoue S.Ring-Opening Polymerizations of Lactone and Epoxide Initiated with Aluminum Complexes of Substituted Tetraphenylporphyrins.Molecular Design of Highly Active Initiators[J].Macromolecules,1990(23):2869.
[19]黄梅,陈关喜.环氧乙烷、环氧丙烷的金属叶琳络合体系催化聚合[J].浙江大学学报,2000,34(3):297-300.
[20]Herold R J.Method of Making a Polyether Using a Double Metal Cyanide Complex Compound:US,3278459[P].1966.
[21]Harper S D.Preparation of Filter Double Metal Cyanide Complex Catalyst for Propylene Oxide Polymerization:EP,0283148[P].1988.
[22]Van der Hulst H,Pogany GA,Kuyper J.Catalysts for the Polymerization of Epoxides and Process for the Prearation of Such Catalysts:US,4477589[P].1984.
[23]Takeyasu HT,Watabe T,Doi T.Production of Polyether: JP,2265921[P].1990.
[24]Jack M.Method of Making a Polyether Using a Double Metal Cyanide Complex Compound:US,3278457[P].1966.
[25]Schuchardt J L,Harper SD.Proceedings of the SPI-32nd Annual Technical/Marketing Conference[C].Pennsylvania:Arco Chem ical Company,1989:360-384.
[26]Robert JH.Polyethers and Method for Making the Same: US,3829505[P].1974.
[27]刘晓华,亢茂青,王心葵.锌/钴双金属氰化物络合物催化(DMC)催化环氧丙烷聚合反应的活性结构研究[J].高等学校化学学报,2000,21(11):1748-1750.
[28]黄亦军,戚国荣,封麟先.双金属氰化物络合物催化环氧烷烃开环聚合的特征[J].高分子学报,2002(3):271-276.
[29]周立宏,唐辉.环氧化物开环聚合催化体系的研究进展[J].精细与专用化学品,2004,12(13):3-5.
[30]BrauneW,Okuda J.An Efficient Method for Controlled Propylene Oxide Polymerization:The Significance of Bimetallic Activation in Aluminum Lewis Acids[J].Angew Chem Int Ed,2003(42):64-68.
[31]Nobori T,Suzuki T,Kiyono S.Phosphazenium Salt and Preparation Process Thereof and Process for Producing Poly (A lkylene Oxide):EP,0791600[P].1997.
[32]于剑昆.磷腈类催化剂及其在聚醚多元醇合成中的应用[J].聚氨酯工业,2004,19(5):10-14.
[33]Zheng Y S,Ying LQ,Shen ZQ.Polymerization of Propylene Oxide by a New Neodymium Complex of Calixanrene Derivative[J].Polymer,2000,41(4):1641-1643.
[34]Jacquier-Gonod V,Llauro M F,Hamaide T.Aluminium and Rare Earths Alkoxides as Initiators for the Heterogeneous Anionic Coordinated Polymerisation of Propylene Oxide.A 1H NMR Approach of the Regioselectivity and Transfer Ability of the Catalytic System[J].Macromol Chem Phys,2000,201(1):12-20.
[35]Nomura R,Wada Y,Haruo H.Polymerization of Oxiranes by Polymeric Organoantimony Oxides[J].J Polymer Sci,Part A:Polymer Chem,1988,26(2):627-636.
Survey of Preparation of Polyalkylene Glycol(PAG)
CHENG Liang
(PetroChina Dalian Lubricating Oil R&D Institute,Dalian 116032,China)
Polyalkylene glycol(PAG)and its applications in lubricants are introduced.The historical background,preparationmethods and polymerizationmechanism of PAG are summarized.Thereinto,preparationmethods of PAG aremainly discussed,including cationic initiation of polymerization,anionic initiation of polymerization,and coordinate-initiated polymerization.And then themechanism and characteristics of each polymerizationmethod and research status of catalysts are summarized.
polyalkylene glycol;lubrication;cationic initiation of polymerization;anionic initiation of polymerization;coordinate-initiated polymerization
TE624.82
A
2011-10-25。
程亮(1981-),男,工程师,2010年毕业于四川大学化学学院有机化学专业,现从事润滑油添加剂的研发工作。
