Phylogenetic evaluation of the taxonomic status of Papilio maackii and P. syfanius (Lepidoptera: Papilionidae)
2011-12-25ZHULiXinWUXiaoBing
ZHU Li-Xin, WU Xiao-Bing
(1. College of Life Sciences, Anhui Normal University, Wuhu Anhui 241000, China; 2. Department of Biology, Chuzhou College, Chuzhou Anhui 239012, China)
Phylogenetic evaluation of the taxonomic status ofPapilio maackiiandP. syfanius(Lepidoptera: Papilionidae)
ZHU Li-Xin1,2, WU Xiao-Bing1,*
(1.College of Life Sciences,Anhui Normal University,Wuhu Anhui241000,China; 2.Department of Biology,Chuzhou College, Chuzhou Anhui239012,China)
The taxonomic status ofPapilio maackiiandP. syfaniushas long been disputed. We conducted a molecular phylogenetic study to evaluate the taxonomic status ofP. maackiiandP. syfanius. A total of twenty-fourP. maackiiindividuals from six localities and sixteenP. syfaniusindividuals from two localities were analyzed. We sequenced the partial region of the CO-I gene (about 579 bp) and partial CO-II gene sequence (about 655bp) of the two species. The Kimura-2-Parameter distances amongP. maackiiandP. syfaniusranged from 0 to 0.6%. Fifteen haplotypes were obtained based on the combined data set. The results strongly supported that allP. maackiiindividuals and allP. syfaniusindividuals formed a large clade, and could not be divided into separated clades. This research indicated that the two species have only very recently undergone speciation.
Papilio maackii;Papilio syfanius; CO-I; CO-II; Phylogeny; Taxonomy
Butterflies of thePapiliogenus are one of the best known invertebrate organisms. They are large and colorful, and particularly noticeable in their habitats. About 210Papiliospecies have been documented worldwide, with some 27 species recorded in China (Wu, 2001).This wide-spread distribution has seen manyPapiliospecies used as model organisms for studies in evolutionary biology, ecology, genetics, and conservation biology (Collins & Morris, 1985; Scriber et al, 1995).
The taxonomic status of severalPrincepsspecies, a subgenus ofPapilio, has long been disputed. SometimesP. maackii, P. dialisandP. syfaniusare treated as subspecies ofP. bianor(Seitz, 1906), yet morphological differences betweenP. bianor,P. maackiiandP. syfaniusare often regarded as sufficient to treat them as separate species. These external morphological characteristics ofP. maackiiandP. syfaniusare as follow:P. maackiiagrees withP. bianorin the character of sexual marks, but differs somewhat in color. Primaries black, thickly powdered with green scales and traversedby a submarginal band formed of paler green scales. Secondaries black, costal area suffused with blue, inner and median area; submarginal lunulated band bluish green, and this color is projected along the third median nervule almost to the extremity of the tail, this is a more or less complete reddish ring at anal angle, outer margin sinuate, fringes white as inP. bianor.P. syfaniusis only separable fromP. bianorby its narrower secondaries, the entire absence of bluish color on the costal area and the presence of a more or less well-defined pale patch on the disc of these wings. This patch is placed at the end of the discoidal cell and is separated into three portions by the discoidal and upper discocellar nervules. These three insects are separated by well-defined and constant characteristics.
TheP. maackiipopulations occur in all over China (except in the north-west), Japan, Korea, and Russia, whileP. syfaniuspopulations occur only in Sichuan, Yunnan, and Xizang. The area inhabited byP. syfaniusis sympatric to the South China distribution ofP. maackii. The host plants ofP. maackiiare Rutaceae, but the biology ofP. syfaniusremains unclear (Wu, 2001).
We examined a large number of these two species from different locations in China. We found that the white patch ofP. syfaniuswas occasionally indistinct, even absent, and that the bluish green band on theP. maackiiindividuals from Southwest China was also sometimes absent. Wing markings onP. maackiiandP. syfaniuswere similar in Southwest China, where the two species were sympatric (Wu, 2001).
Despite the extensive use ofPapiliospecies in basic research, the phylogeny ofPapilioremains weak. Recent relevant studies have used mitochondrial DNA (mtDNA) as molecular markers (Sperling & Harrison, 1994; Aubert et al, 1999; Reed & Sperling, 1999; Yagi et al, 1999; Caterino & Sperling, 1999; Caterino et al,2001; Zakharov et al, 2004a, b; Zhu et al, 2007; Silva-Brandão et al, 2008; Wheata & Watt, 2008; Chen et al, 2010), as mtDNA is considered useful in phylogenetic studies due to its rapid evolution. However,P. maackiiandP. syfaniuswere only distinguished by less pairwise substitutions (0.15%) based on partial mitochondrial COI and CO-II gene sequences. The sequences of their CO-I and CO-II gene fragments were the same, although sequence divergences among these two species and other Chinese species ofPrincepsranged from 3.6% (P. polyctor) to 8.28% (P. demoleus), and sequence divergences among them andP. dialiswere 4.33% (Zhu et al, 2007). These results indicated thatP. maackiiandP. syfaniuscould be conspecific.
So, isP. syfaniusa synonym species ofP. maackii? Further evidence was required to answer this question. Previous research determined that DNA sequencing of a standard gene region of CO-I gene or “DNA barcoding”(Hebert et al, 2003) might hold the answer. DNA barcoding is helpful in species diagnosis because sequence divergences are ordinarily much lower among individuals of a species than between closely related species (Hebert et al, 2003, 2004a, b; Avise & Walker, 1999). To provide further proof, we sequenced the standard gene region of the CO-I gene or “DNA bar coding” and partial CO-II gene sequence of the two species. We focused on and attempted to evaluate the taxonomic status ofP. maackiiandP. syfanius.
1 Materials and Methods
1.1 Insects
We obtained twenty-four adult individuals ofP. maackiifrom six localities in China and sixteen adult individuals ofP. syfaniusfrom two localities in South China, where the two species were sympatric (Fig. 1; Tab. 1). These specimens included both fresh and papered specimens. The sample ofP. bianorwas obtained for use as an out-group. Specimens were either collected by us or donated by colleagues. Sequences from all specimens were available from GenBank (Tab. 1).
1.2 DNA extraction
Tissue samples were comprised of two legs removed from one side of the thorax to preserve both the taxonomic and aesthetic values of the specimens. Voucher labels were attached to all sampled specimens. Excised legs from dried materials were rehydrated in buffer consisting of 5 mmol/L of Tris-HCl, 25 mmol/L of NaCl, pH 8.0, 25 of mmol/L EDTA, and 0.1% SDS (Zimmermann et al, 2000) for at least 48 h. A total of 5 µL µL of 20 mg/mL Proteinase K enzyme digester was added, and samples were incubated at 55°C overnight. Total DNA was extracted sequentially with phenol, phenol/chloroform (1/1), and chloroform and then precipitated with ethanol. The resultant DNA was dissolved in 30 µL ul of double-distilled H2O.
1.3 Amplification and sequencing of DNA
Total DNAs were used as templates for amplifications of the partial CO-I gene (about 579bp) and CO-II gene (about 655bp) by polymerase chain reaction (PCR). These sequences were amplified by the following primer pairs for Lepidoptera: LEP-F1, 5'-ATTCAACCAATCA TAAAATAT-3'; LEP-R1, 5'-TAAACTTCTGATGTCC AAAAA-3' for the COI gene (Hebert et al, 2004a) and PATRICK, 5'-CTAATAT GGCAGATTATATGTATTG GA-3'; EVA, 5'-GAGACCATTACTTGCTTTCAGTCA CT-3'for the COII gene (Caterino & Sperling, 1999).

Fig. 1 Distribution of sampling sites in China
All PCR mixes had a total volume of 50 µL and contained 10 mmol/L of Tris-HCl (pH 8.3), 50 mmol/L of KCl, 1.5 mmol/L MgCl2, 100 µmol/L of dNTPs, 10-50 ng (1-5µL) of genomic DNA, and 1 UTaqDNA polymerase. The thermocycling profile consisted of one cycle of 1 min at 94°C, 6 cycles of 1 min at 94°C, 1 min and 30 sec at 48°C, and 1 min and 15 sec at 72°C, followed by 36 cycles of 1 min at 94°C, 1 min and 30 sec at 52°C, and 1 min and 15 sec at 72°C, and a final step of 5 min at 72°C. The PCR products were electrophoresed in 1.0% TBE agarose gels, stained with ethidium bromide, and visualized under UV light. The PCR products were cleaned using the V-Gene DNA Gel Extraction Kit. The same primers for PCR were used for sequencing. Sequences were analyzed on an ABI 3730 automated sequencer.
1.4 Phylogenetic analysis
The sequence data were aligned with CLUSTALX (Thompson et al, 1997) and by eye. All variable characters, parsimony-informative characters, and the Tis/TVs were estimated from pairwise comparisons of sequences with Mega Version 2.1 based on Kimura-2-Parameter (Kumar et al, 2001). The ML analyses were performed using the equally weighted sequence data with standard settings with PAUP* Version 4.0b10 (Swofford, 1998). The GTR+G+I model was calculated by Modeltest 3.06 (Posada & Crandall, 1998). The statistical confidence for each clade was determined using the bootstrap test based on 100 replicates for ML using PAUP*. MrBayes 3.04b (Ronquist & Huelsenbeck, 2003) was used for the Bayesian estimation of phylogeny. The GTR+I+G model was used for Bayesian inference. An initial run was performed to determine the burn-in value, which was used in further analyses to exclude all trees prior to the stable log likelihood estimate. Searches were conducted with four simultaneous Markov Chains over two million generations, sampling every 100 generations. To estimate the posterior probabilities of recovered branches, the 50% majority rule was applied. To ensure that Bayesian inference was not trapped in local optima, the analysis was performed three times, starting from different random trees. Phylograms were created as average-branch length consensus trees in MrBayes and visualized with Tree-View 1.6.6 (Page, 1996). Bootstrap values were shown above the node. A partition homogeneity test was carried out in PAUP* to determine if significantly different signals were being generated by the CO-I and CO-II fragments. To further resolve relationships within major haplotype clusters, a statistical parsimony approach was employed (Templeton et al, 1992), which was carried out using computer program TCS 1.18 (Clement et al, 2000).
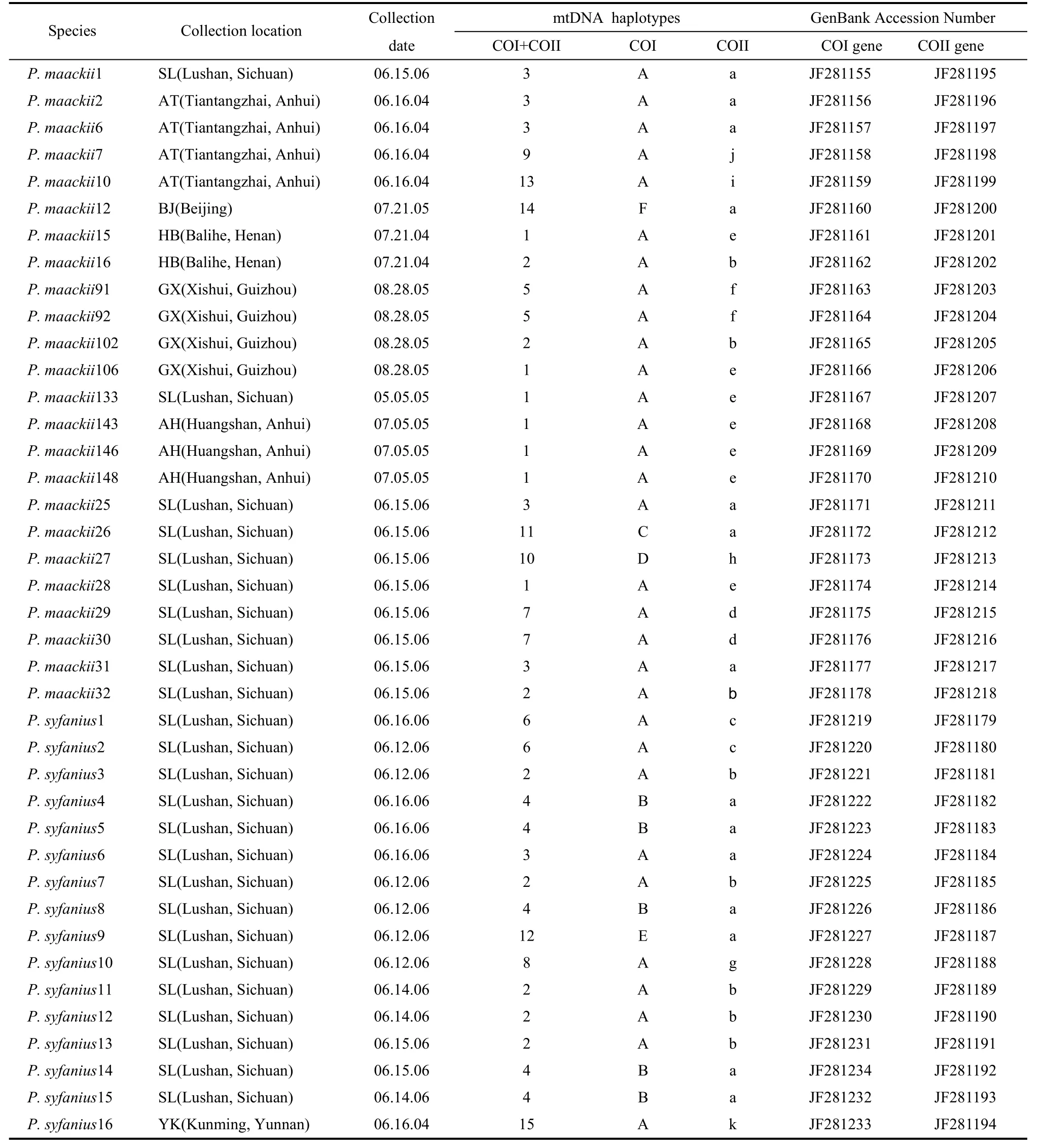
Tab. 1 Specimens, collecting localities and dates, mtDNA haplotypes and the NCBI accession codes for the Papilio mtDNA sequences
2 Results
Sequences used to generate phylogeny represented about 7.7% of the insect mitochondrial molecule. Of the 1234 characters (579bp of COI and 655bp of CO II) in the data matrix, nineteen (1.54%) were variable and eight (0.65%) were parsimony informative. The partition homogeneity test showed no significant incongruence between phylogenetic signals from CO-I and CO-II fragments (P=0.38). The average transition/transversion ratio was calculated as 4.8. The Kimura-2-Parameter distances amongP. maackiiandP. syfaniusranged from 0 to 0.6%, with an overall average of 0.2%.
These sequences produced a combined data set of fifteen haplotypes (Tab. 1). Haplotypes 2 and 3 were shared by the two species. This was particularly true for the haplotypes produced by the CO-I gene and CO-II gene region data (Tab. 1). The CO-I region data included six unique haplotypes, and the CO-II region data included eleven haplotypes. Haplotypes A of the CO-I region and haplotypes A and B of the CO-II region were shared byP. maackiiandP. syfanius. In both the parsimony and Bayesian analyses,P. bianorwas used as the out-group. The Bayesian majority consensus tree for the combined data set is presented in Fig. 2. The ML tree had a nearly identical topology to the majority consensus tree of the Bayesian inference. Most nodes were supported by both methods. Results showed that allP. maackiiandP. syfaniusindividuals formed a large clade, and could not be divided into separated clades. These groups corresponded closely to the distribution of the major clades defined by TCS.
The statistical parsimony analysis of the combined data set produced one network, which contained all of the haplotypes (Fig. 3.). The networks provided the relationships of the haplotypes. Haplotype 2 and 3 were central and by far the most numerous in this network (Tab. 1). The network was also unable to separateP. syfaniusfromP. maackii.
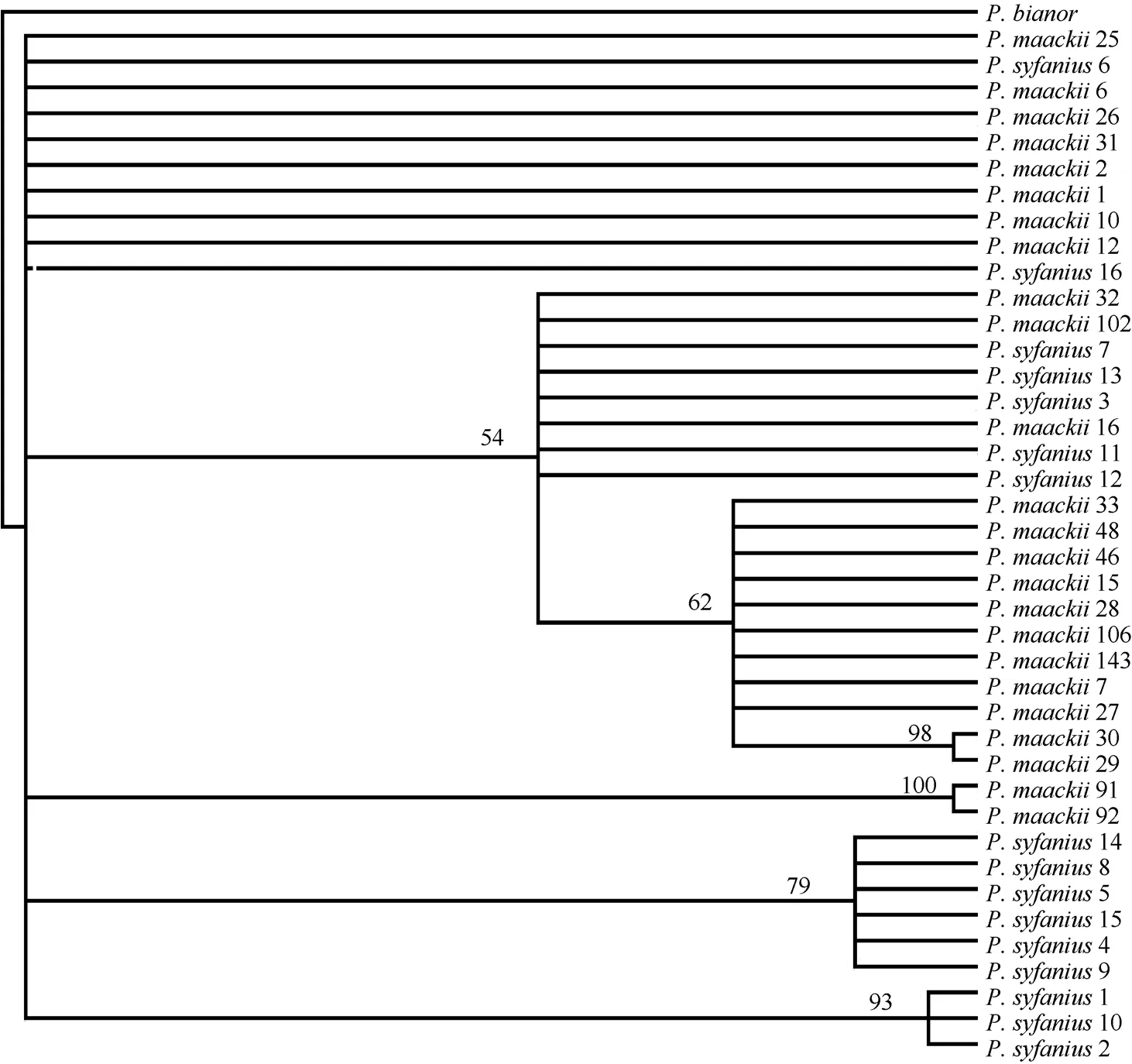
Fig. 2 Bayesian majority consensus tree of Papilio maackii and P. syfanius based on sequences of CO-I (579 bp) and CO-II (655 bp)
3 Discussion
We examined the phylogeny derived from concatenated CO-I and CO-II gene fragments ofP. maackiiandP. syfaniusfrom China, and provided an insight into their taxonomic status.
The taxonomic status ofP. maackiiandP. syfaniushas long been disputed. DNA barcoding can be helpful in species diagnosis because sequence divergences are ordinarily much lower among individuals of a species than between closely related species (Hebert et al, 2003, 2004a, b; Avise & Walker, 1999). For example, con-generic moth species show an average sequence divergence of 6.5% in the CO-I gene, whereas divergences among con-specific individuals averages only 0.25% (Moore, 1995). Similar values were obtained in birds, with intra-specific divergences at CO-I averaging 0.27%, whereas congener divergences averaged 7.93% (Hebert et al, 2003, 2004a, b; Avise & Walker, 1999).Princepsdivergences we obtained previously ranged from 3.6% to 8.28% (Zhu et al, 2007). To provide further proof, we sequenced partial CO-II gene sequences of the two species. Based on previous studies of butterflies, con-generic species show an average sequence divergence of 4% in CO-II gene (Sperling et al, 1996; Wang et al, 2004), whereas divergences among conspecific individuals average 1%-2% (Sperling & Hickey, 1994). We sequenced the standard gene region of the CO-I gene (about 579bp), known as “DNA barcoding”, and the partial CO-II gene sequence (about 655bp) of the two species.
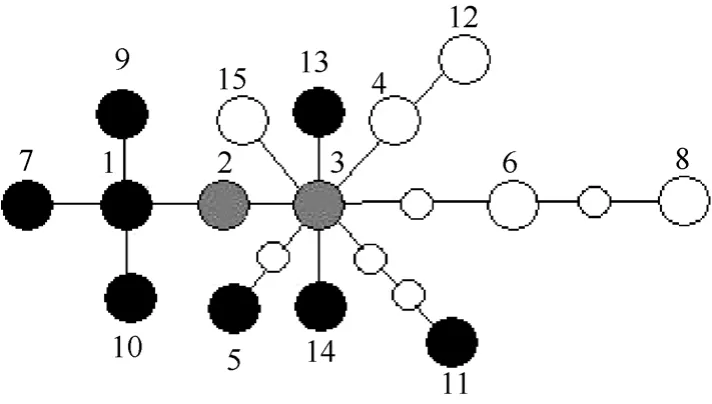
Fig. 3 Statistical parsimony network of haplotypes of Papilio maackii (black circles), P. syfanius (white circles), and both species (gray circles)
Following Mayr’s (1963) biological species concept, it is generally assumed that species must be monophyletic entities (Harrison, 1998). However, as shown by Pamilo & Nei (1988) and summarized by Wahlberg et al (2003) and Funk & Omland (2003) both polyphyly and paraphyly may arise during the speciation process. Although what constitutes a species is a subject of intense debate, there is a general agreement that species are segments of evolutionary lineages (de Queiroz, 1998).
Our results indicated that sequence divergence betweenP. maackiiandP. syfaniusranged from 0 to 0.6% across both CO-I and CO-II gene fragments. The phylogenetic analysis could not separateP. maackiiandP. syfaniusinto different clades, instead supporting a monophyly (Fig.2). The clade included all theP. maackiiandP. syfaniusindividuals, with neither species able to form separated clades and both sharing the same haplotypes. If we were unfamiliar withPapiliobutterflies, we could, using the ‘‘classic” 665 bp barcoding segment of CO-I (Hebert et al, 2004a), place the two specimens into their ‘‘correct” major subclades and sometimes into the ‘‘correct” species. They would have genetic distinctness of geographic subgroups of a morphologically conservative assemblage previously called one species. But the phylogeny of many subclades is poorly resolved with this short sequence block, and recent divergences, clear when more diverse signal sources are used, would go undetected. ‘‘Barcoding” is useful for the initial sorting of material, but it is no substitute for phylogenetic studies using more extensive evidence.
Results from the probabilistic modeling approach indicate that accurate species delimitation is possible (Knowles & Carstens, 2007), despite widespread incomplete lineage sorting and discordance among loci, and confirm that it is not necessary to rely on exclusivity criteria. Molecular data may suggest that morphological differentiation proceeded much faster than mtDNA divergence owing to strong selection pressures (Eastwood & Hughes, 2003), sinceAcrodipsaswere allopatric and lived in markedly different habitats. This fixed amino acid change may be a “key innovation” of macroevolutionary consequences, as the derived genotypes carrying it in the warm habitats of the lowland complex, produce much more thermally stable PGI than the basal ones in colder, higher or more northern habitats (Christopher & Ward, 2008). Studies on the intermediate taxa in this phylogeny, in regards to historical relationships and the state of adaptations, may illuminate connections between micro- and macro-evolutionary processes. Our results indicate that the two species have undergone only very recent speciation.
Aubert J, Legal L, Descimon H, Michel F. 1999. Molecular phylogeny of swallowtail butterflies of the tribe Papilionini (Papilionidae, Lepidoptera) [J].Mol Phyl Evol, 12:156-167.
Avise JC, Walker D. 1999. Species realities and numbers in sexual vertebrates: Perspectives from an asexually transmitted genome [J].Proc Nat Acad Sci USA, 96: 992-995.
Caterino MS, Sperling FAH. 1999.Papiliophylogeny based on mitochondrial cytochrome oxidase I and II genes [J].Mol Phyl Evol, 11: 122-137.
Caterino MS, Reed RD, Kuo MM, Sperling FAH. 2001. A partitioned maximum likelihood analysis of swallowtail butterfly phylogeny (Lepidoptera: Papilionidae) [J].Syst Biol, 50: 106-127.
Chen Jun, Li Qi, Kong Ling-Feng, Zheng Xiao-Dong, Yu Rui-Hai. 2010. COI-based DNA barcoding in Tapetinae species (Mollusca, Bivalvia, Veneridae) along the coast of China[J].Zool Res, 31(4): 345-352.
Christopher WW, Ward BW. 2008. A mitochondrial-DNA-based phylogeny for some evolutionary-genetic model species ofColiasbutterflies (Lepidoptera, Pieridae) [J].Mol Phyl Evol, 47: 893-902.
Clement M, Posada D, Crandall K. 2000. TCS: a computer program to estimate gene genealogies [J].Mol Ecol, 9:1657-1660.
Collins NM, Morris MG. 1985. Threatened Swallowtail Butterflies of the World [M]. The IUCN Red Data Book. IUCN. UK: Cambridge.
de Queiroz K. 1998. The general lineage concept of species, species criteria, and the process of speciation: A conceptual unification and terminological recommendations[M]// Howard DJ, Berlocher SH. Endless Form, Species and Speciation. New York: Oxford University Press, 57-75.
Eastwood R, Hughes JM. 2003. Molecular phylogeny and evolutionary biology ofAcrodipsas(Lepidoptera : Lycaenidae ) [J].Mol Phyl Evol, 27: 93-102.
Funk DJ, Omland KE. 2003. Species-level paraphyly and polyphyly: Frequency, causes, and consequences, with insights from animal mitochondrial DNA [J].Annu Rev Ecol Evol Syst, 34: 397-423.
Harrison RG. 1998. Linking evolutionary pattern and process: the relevance of pecies concepts for the study of speciation[M]// Howard DJ & Berlocher SH. Endless Forms: Species and Speciation. Oxford: Oxford University Press.19-31, 470.
Hebert PDN, Cywinska A, Ball SL, Waard de JR. 2003. Biological identification through DNA barcodes [J].Proc Roy Soc B, 270: 313-321.
Hebert PDN, Penton EH, Burns JM, Janzen DH, Hallwachs W. 2004a. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterflyAstraptes fulgerator[J].Proc Nat Acad Sci USA, 101: 14812-14817.
Hebert PDN, Stoeckle MY, Zemlak TS, Francis CM. 2004b. Identification of birds through DNA barcodes [J].PLoS Biol, 2: 1657-1663.
Knowles LL, Carstens BC. 2007. Delimiting species without monophyletic gene trees [J].Syst Biol, 56: 887-895.
Kumar S, Tamura K, Jakobsen IB, Nei M. 2001. MEGA2: Molecular Evolutionary Genetics Analysis Software [M]. Arizona State University, Tempe, AZ.
Mayr E. 1963: Animal Species and Evolution [M]. Cambridge, Mass: Belknap Press.
Moore WS. 1995. Inferring phylogenies from mtDNA variation: Mitochondrial-gene trees versus nuclear-gene trees [J].Evolution, 49: 718-729.
Page RD. 1996.TreeView: an application to display phylogenetic trees on personal computers [J].Comput Appl Biosci, 12: 357-358.
Pamilo P, Nei M. 1988: Relationships between gene trees and species trees [J].Mol Phyl Evol, 5: 568-583.
Posada D, Crandall KA. 1998. Modeltest: testing the model of DNA sustitution [J].Bioinfomatics, 14: 817-818.
Reed RD, Sperling FAH. 1999. Interaction of process partitions in phylogenetic analysis: An example from the swallowtail butterfly genusPapilio[J].Mol Phyl Evol, 16: 286-297.
Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models [J].Bioinformatics, 19: 1572-1574.
Seitz A. 1906. The Macrolepidoptera of the World. Stuttgart: A [M]. Kernen. vol.1: 10.
Scriber JM, Tsubaki Y, Lederhouse RC. 1995. Swallowtail Butterflies: Their Ecology and Evolutionary Biology [M]. Florida: Scientific Publishers Gainesville.
Silva-Brandão KL, Wahlberg N, Francini RB, Azeredo-Espin AML, Brown KSJ, Paluch M, Lees DC, Freitas AVL. 2008. Phylogenetic relationships of butterflies of the tribe Acraeini (Lepidoptera, Nymphalidae, Heliconiinae) and the evolution of host plant use [J].Mol Phyl Evol, 46(2): 515-31
Sperling FAH, Harrison RG. 1994. Mitochondrial DNA variation within and between species of thePapilio machaongroup of swallowtail butterflies [J].Evolution, 48: 408-422.
Sperling FAH, Hickey DA. 1994. Mitochondrial DNA sequence variation in the spruce budworm species complex (Choristoneura: Lepidoptera) [J].Mol Phyl Evol, 11: 656-665.
Sperling FAH, Landry JF, Hickey D. 1996. Mitochondrial DNA sequence variation among pheromotypes of the dingy cutworm,Feltia jaculifera(Lepidoptera : Noctuidae) [J].Can J Zool, 74: 2 109-2 117.
Swofford DL. 1998. PAUP: Phylogenetic Analysis Using Parsimony (and other methods). Version 4 [M]. Sunderland, Massachusetts: Sinauer Associates.
Templeton AR, Crandall KA, Sing CF. 1992. A cladistic analysis of phenotypic association with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation [J].Genetics, 132: 619-633.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The clustalx windows interface: exible strategies for multiple sequence alignment aided by quality analysis tools [J].Nucleic Acids Res, 24: 4 876-4 882.
Wahlberg N, Oliveira R, Scott JA. 2003. Phylogenetic relationships ofPhyciodesbutterfly species (Lepidoptera: Nymphalidae): complex mtDNA variation and species delimitations [J].Syst Entomol,28: 257-273.
Wheata CW, Watt WB. 2008. A mitochondrial-DNA-based phylogeny for some evolutionary-genetic model species of Colias butterflies (Lepidoptera, Pieridae) [J].Mol Phyl Evol, 47(3): 893-902.
Wu CS. 2001. Fauna Sinica Insect Vol.25 Lepidoptera Papilionidae [M]. Beijing: Science Press.
Yagi T, Sasaki G, Takebe H. 1999. Phylogeny of Japanese papilionid butterflies inferred fromnucleotide sequences of the mitochondrial ND5 gene [J].J Mol Evol, 48: 42-48.
Zakharov EV, Caterino MS, Sperling FAH. 2004a. Molecular phylogeny, historical biogeography, and divergence time estimates for swallowtail butterflies of the genusPapilio(Lepidoptera: Papilionidae) [J].Syst Biol, 53: 193-215.
Zakharov EV, Campbell RS, David CL, Cameron A, Vane-Wright RI, Sperling FAH. 2004b. Independent gene phylogenies and morphology demonstrate a malagasy origin for a wide-ranging group of swallowtail butterflies [J].Evolution, 58: 2763-2782.
Zimmermann M, Wahlberg N, Descimon N. 2000. Phylogeny of Euphydryas checkspot butterflies (Lepidoptera: Nymphalidae) based on mitochondrial DNA sequence data [J].Ann Entomol Soc Am, 93: 347-335.
Zhu LX, Wu XB, Wu CS. 2007. Phylogeny ofPrincepsbutterflies from China: evidences from mitochondrial partial COI and COII gene sequences (Lepidoptera: Papiliondae) [J].Curr Zool, 53(2): 257-263.
基于系统发生探讨绿带翠凤蝶和西番翠凤蝶的分类地位
诸立新1,2, 吴孝兵1,*
(1. 安徽师范大学 生命科学院, 安徽 芜湖 241000; 2. 滁州学院生物系, 安徽 滁州 239000)
绿带翠凤蝶和西番翠凤蝶的分类问题存在一定的争议。应用分子系统学方法对这一问题进行了研究。对6个不同地区的24个绿带翠凤蝶、2个地区的16个西番翠凤蝶个体的 COI (579bp)和 COII (655bp)基因测序,绿带翠凤蝶与西番翠凤蝶的遗传距离为 0至0.6%, 共获得了 15个单倍型。结果显示这些单倍型不能形成各自独立的单系群, 因此认为绿带翠凤蝶和西番翠凤蝶为近期分化的两个种。
绿带翠凤蝶; 西番翠凤蝶; CO-I; CO-II; 系统发生; 分类
Q969.451.8; Q349.1; Q951.3
A
0254-5853-(2011)03-0248-07
2010-11-01;接受日期:2011-04-18
10.3724/SP.J.1141.2011.03248
date: 2010-11-01; Accepted date: 2011-04-18
s: National Natural Science Foundation of China (30870359); Anhui Scholar Special Fund in Science and Technology (KJ2009B015)
*Corresponding author (通讯作者), Tel: 86-0553-3836873, Fax: 86-0553-3836873, E-mail: wuxb@mail.ahnu.edu.cn
