吡咯并[2,3-d]嘧啶衍生物的合成方法研究进展*
2011-11-26袁京莉史博颖曹胜利
袁京莉, 史博颖, 鲁 丰, 曹胜利
(首都师范大学 化学系,北京 100048)
吡咯并[2,3-d]嘧啶是重要的杂环化合物母体。近年来,以2,4-二氨基吡咯并[2,3-d]嘧啶或2-氨基-4-氧代吡咯并[2,3-d]嘧啶为母体的经典或非经典抗叶酸剂的合成及生物活性研究,在抗癌药物研究中占有重要的位置[1,2]。文献报道的这两类化合物的合成路线较多,所用的原料和反应条件也有较大差别。在合成2,4-二氨基吡咯并[2,3-d]嘧啶类化合物(Ⅰ, Chart 1)的文献中,大多是先合成2-氨基-3-氰基呋喃类衍生物,再与胍反应形成吡咯并嘧啶类化合物;也有以吡咯、2,4,6-三氨基嘧啶或者2-氨基-4-氧代吡咯并[2,3-d]嘧啶为原料来合成的报道。而合成2-氨基-4-氧代吡咯并[2,3-d]嘧啶类化合物(Ⅱ, Chart 1)则大多以2,6-二氨基-4-氧代嘧啶为原料,再形成吡咯环。本文归纳总结了这两类化合物的主要合成方法,并对各种方法的优缺点进行简要评价。
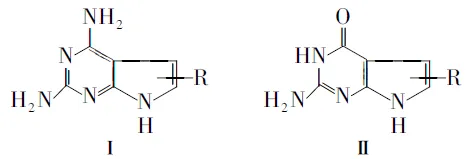
Chart1
1 Ⅰ的合成
1.1 以呋喃衍生物为原料或中间体
Taylor等[3]将4-位取代的2-氨基-3-氰基呋喃与游离的胍(用等摩尔比的NaOMe调节盐酸胍得来)在乙醇中回流反应24 h后,再用乙酸酸化反应液得5-位取代的2,4-二氨基吡咯并[2,3-d]嘧啶(1~3, Scheme 1),收率分别为67%, 66%和42%。此方法条件温和,收率也较高。
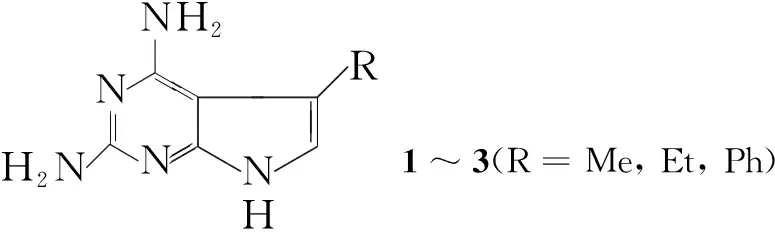
Scheme1
Scheme2
Scheme3
Scheme4
Gangjee等[4,5]以甲醇为溶剂,在三乙胺存在下,将羟基丙酮与丙二腈在室温下搅拌过夜制得2-氨基-3-氰基-4-甲基呋喃。再与盐酸胍在甲醇钠的乙醇溶液中回流反应24 h合成了1(Scheme 2),两步收率48%。此方法省去了乙酸调节pH值的步骤,比Taylor的方法操作简单。Gangjee等[6]又用此法,以1-羟基-2-丁酮为原料,合成了2(Scheme 2),第二步收率为66%。
Gangjee等[7]将丙二腈和三乙胺溶于无水甲醇中,向反应液中滴加1-羟基-2-戊酮,室温搅拌24 h,减压蒸除溶剂后经柱色谱法提纯得到2-氨基-3-氰基-4-丙基呋喃,收率60%。再将其加到盐酸胍、甲醇钠的乙醇溶液中,回流反应过夜;减压蒸除溶剂后经柱色谱法提纯得到2,4-二氨基-5-丙基吡咯并[2,3-d]嘧啶(4, Scheme 3),收率50%。利用上述方法,还合成了2,4-二氨基-5-异丙基吡咯并[2,3-d]嘧啶(5, Scheme 3),两步总收率为44%。
Rosowsky等[8,9]将丙二腈和三乙胺溶于无水甲醇中,向反应液中滴加1-羟基-2-苯基乙酮或1-羟基-3-芳基丙酮,室温反应24 h得到4-位取代的2-氨基-3-氰基呋喃,收率40%~60%。将此中间体加到盐酸胍、甲醇钠的乙醇溶液中,回流反应过夜得到5-位取代的2,4-二氨基吡咯并[2,3-d]嘧啶(6, Scheme 4),收率30%~60%。Gangjee等[10]用相似的方法,将反应原料改为5-芳基取代的1-羟基-2-戊酮,得到2,4-二氨基-5-(3-芳基丙基)吡咯并[2,3-d]嘧啶(6, Scheme 4),收率50%~90%。
Edward等[11]使用与Gangjee等[4,5]类似的方法,将4-[3-(5-氨基-4-氰基呋喃)丙基]苯甲酸乙酯与盐酸胍反应、甲醇钠溶于乙醇中,回流反应24 h得到4-[3-(2,4-二氨基吡咯并[2,3-d]嘧啶-5-)]丙基苯甲酸乙酯(7, Scheme 5),收率44%。
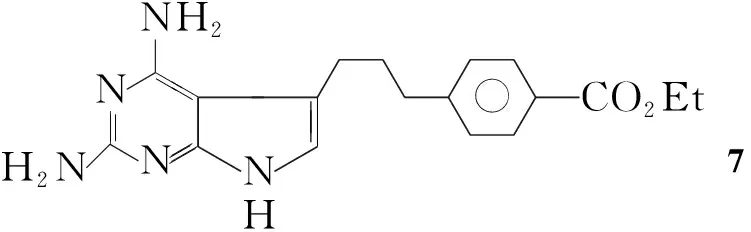
Scheme5
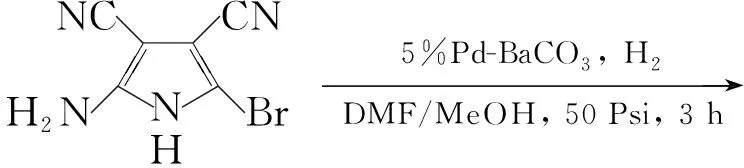
Scheme6

Scheme7
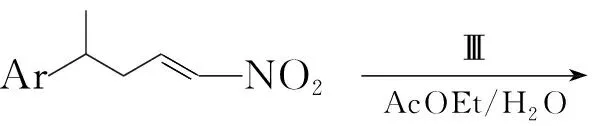
Scheme8
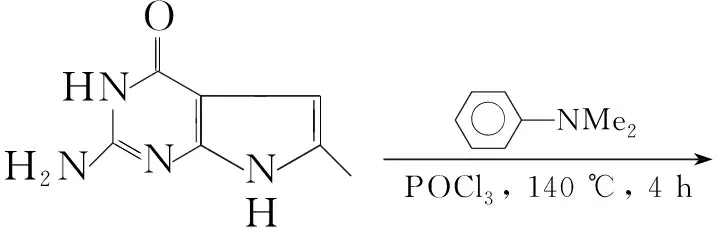
Scheme9
1.2 以吡咯衍生物为原料
Gangjee等[12]将2-氨基-3,4-二氰基-5-溴吡咯和5%Pd-BaCO3溶于DMF和甲醇中,在50 Psi下氢化3 h得到2-氨基-3,4-二氰基吡咯(A),收率64%。以Dowtherm-A为溶剂,将A与氯甲脒盐酸盐反应,于160 ℃~170 ℃反应48 h得到2,4-二氨基-5-氰基吡咯并[2,3-d]嘧啶(8, Scheme 6),粗品收率79%。 8不经纯化直接用于下步反应。此方法反应时间较长,反应条件比较苛刻,但产率较高。他们又将2-氨基-3,4-二氰基呋喃在乙醇钠存在下与盐酸胍反应,但没有得到预期的产物。
1.3 以2,4,6-三氨基嘧啶(Ⅲ)为原料
Zhu等[13]用氯乙腈与甲酸甲酯在以甲醇钠为碱的条件下反应得到2-氯-2-甲酰基乙腈(B),收率60%。将Ⅲ与乙酸钠的水溶液混和,于50℃/1 h内滴加B;于室温反应12 h后再回流反应1 h得8(Scheme 7),收率66%。此方法收率较高,但操作较为复杂。
Gangjee等[14]将1-硝基-4-芳基戊烯与Ⅲ在乙酸乙酯/水中反应16 h得到2,4,6-三氨基-5-(1-硝基-4-芳基-2-戊基)嘧啶(C),收率80%~84%。将C加入2 mol·L-1NaOH(3 mL)中,于室温反应2 h;冷却至0 ℃,加入2.5 mol·L-1H2SO44 mL;在0 ℃下,用2 mol·L-1NaOH调至pH 7;于室温反应1 h后再用冰醋酸酸化。最后经柱色谱法纯化得2,4-二氨基-5-(2-芳基丙基)-吡咯并[2,3-d]嘧啶(9, Scheme 8),收率45%~53%。
1.4 以Ⅱ为原料
Gangjee等[15]以N,N-二甲基苯胺为溶剂,将2-氨基-4-氧代-6-甲基吡咯并[2,3-d]嘧啶与三氯氧磷在140 ℃下反应4 h得到2-氨基-4-氯-6-甲基吡咯并[2,3-d]嘧啶(D),收率32%。冰浴冷却下,将D加入到饱和的氨/甲醇溶液中,于125 ℃~135 ℃反应48 h得到2,4-二氨基-6-甲基吡咯并[2,3-d]嘧啶(10, Scheme 9),收率76%。
Scheme10
最近,Gangjee等[16]将2-氨基-4-氧代-5-氯-3,9-二氢吡啶并[4,5-b]吲哚与2,2-二甲基丙酸酐、DMAP和三乙胺加入到DMF中,于60 ℃反应48 h得到氨基被保护的产物(E),收率40%。将E溶于三氯氧磷中,加热至110 ℃~120 ℃反应4 h得到N-(4,5-二氯吡啶并[4,5-b]吲哚基)-2,2-二甲基丙酰胺(F)。将其加入饱和氨气的甲醇溶液中,于130 ℃反应48 h得到5-氯-吡啶并[4,5-b]吲哚-2,4-二氨基丙酰胺(G)。最后,在微波辐照下,将G和硫酚或对甲基硫酚、碳酸钾、NMP于250 ℃反应30 min得产物11或12(Scheme 10),收率分别为97%, 87%。此方法反应步骤较多,操作复杂,但产率较高。
2 Ⅱ的合成
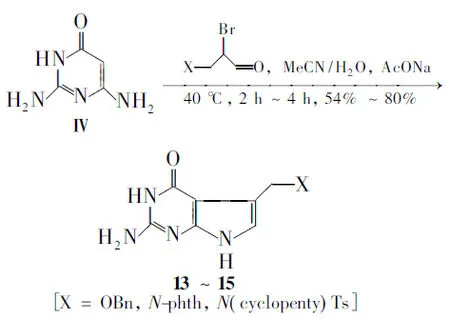
2.1 以2,6-二氨基-4-氧代嘧啶(Ⅳ)为原料
Charles等[17]将Ⅳ, C3-位取代的2-溴丙酮和乙酸钠溶于体积比为1∶1的乙腈/水溶液中,于40 ℃反应2 h~4 h得到5-取代的2-氨基-4-氧代吡咯并[2,3-d]嘧啶类衍生物(13~15, Scheme 11),收率50%~80%。此方法反应条件温和,收率较好。
Scheme11
Gangjee等[18]采用类似的方法,将Ⅳ与2-氯-2-甲酰基乙腈或2-氯-2-甲酰基乙酸甲酯加入到乙酸钠水溶液中,回流反应5 h得到2-氨基-4-氧代-5-氰基吡咯并[2,3-d]嘧啶(16)或2-氨基-4-氧代吡咯并[2,3-d]嘧啶-5-甲酸甲酯(17, Scheme 12),收率分别为72%, 62%。
Scheme12
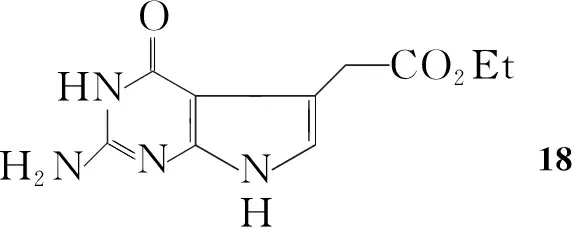
Gangjee等[19]将Ⅳ加入到乙酸钠的水溶液中,加热至回流后滴加4-氯乙酰基乙酸乙酯,滴毕,继续反应18 h得到2-氨基-4-氧代-6-乙酸甲酯基吡咯并[2,3-d]嘧啶(18, Scheme 13),收率54%。
Scheme13
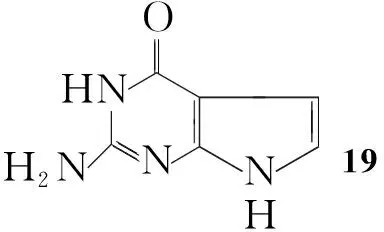
Gangjee等[20]将Ⅳ与氯乙醛加入到乙酸钠的水溶液中,回流反应4 h得到2-氨基-4-氧代吡咯并[2,3-d]嘧啶(19, Scheme 14),收率82%。此方法操作简单,收率较高。
Scheme14
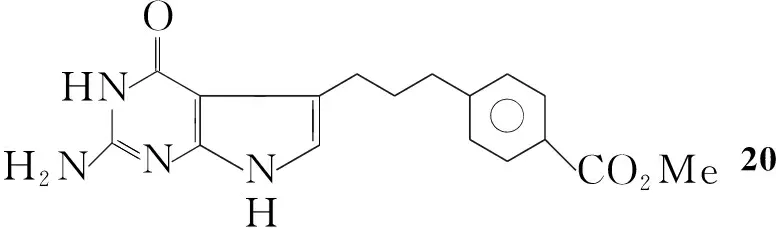
Gangjee等[21]将氯甲基-3-(4′-甲氧苯基)丙酮加入Ⅳ的无水DMF溶液中,于40 ℃~50 ℃反应3 d得到4-[3-(2-氨基-4-氧代-吡咯并[2,3-d]嘧啶基)丙基]苯甲酸甲酯(20, Scheme 15),收率40%。此方法操作简单,但收率较低。
Scheme15
Gangjee等[22]将Ⅳ加入到乙酸钠的水溶液中,于100 ℃加入2-氯-2-甲酰基乙腈,回流反应4 h得到2-氨基-4-氧代-6-甲基吡咯并[2,3-d]嘧啶(21, Scheme 16),收率70%。
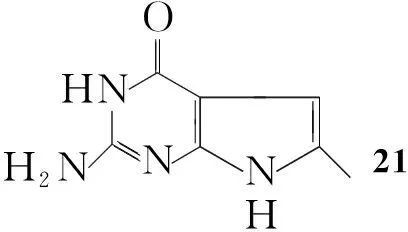
Scheme16
Gangjee等[5]将丙醛溶于二氧六环中,于0 ℃缓慢滴加液溴,反应10 min得2-溴丙醛(H),收率62%。将H和Ⅳ,三乙胺溶于DMSO中,于室温反应1 h后经柱色谱法纯化得到2-氨基-4-氧代-6-甲基吡咯并[2,3-d]嘧啶(22, Scheme 17),收率71%。此方法反应时间短,收率较高。
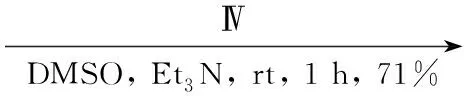
Scheme17
Gangjee等[23]将Ⅳ和1-溴-2-丁酮溶于DMF中,氮气保护下于40 ℃~50 ℃反应72 h。减压蒸除溶剂后经柱色谱法纯化得到2-氨基-4-氧代-6-乙基吡咯并[2,3-d]嘧啶(23, Scheme 18),收率40%。
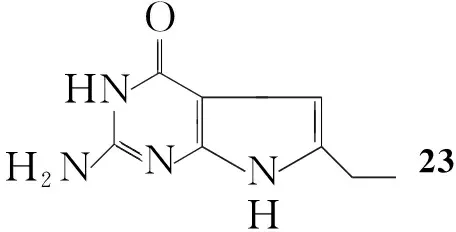
Scheme18
Gangjee等[24]将Ⅳ和4-(3-氯-2-羰基丙基)苯甲酸甲酯溶于DMF中,于50 ℃~60 ℃反应2 d得到4-(2-氨基-4-氧代吡咯并[2,3-d]嘧啶-6-)苯甲酸甲酯(24, Scheme 19),收率60%。
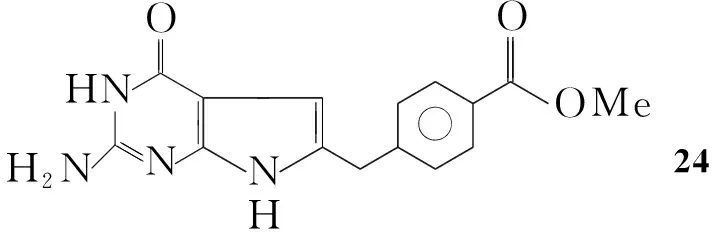
Scheme19
Zhu等[13]用氯乙腈与甲酸甲酯在甲醇钠为碱的条件下合成B,收率60%。将Ⅳ加入乙酸钠水溶液中,于50 ℃滴加B(1 h内)。冷却至室温,搅拌12 h后回流反应1 h得到16(Scheme 20),收率72%。此方法比Gangjee等[5]的方法操作复杂,反应时间长,收率并没有很大提升。
Scheme20
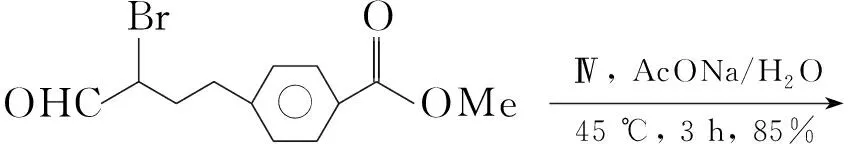
Scheme21
Min等[25]将Ⅳ加入水和甲醇混合液中;加入4-(3-溴-3-甲酰基丙基)苯甲酸甲酯和醋酸钠,于45 ℃反应3 h得产物25(Scheme 21),收率85%。
最近,Gangjee等[26,27]用两种方法合成了21。方法一:将Ⅳ和一氯丙酮溶于DMF中,于60 ℃反应48 h得到21(Scheme 22),但同时产生副产物2,4-二氨基-5-甲基呋喃并[2,3-d]嘧啶。方法二:将Ⅳ加入到乙酸钠的水溶液中,溶液变澄清后加热至100 ℃后快速加入一氯丙酮,反应4 h得21(Scheme 22),收率70%。方法二比方法一反应条件温和,无副产物,收率较高。Tangeda等[28]在方法一的基础上改进,将Ⅳ加入到乙酸钠水溶液中,于80 ℃快速加入一氯丙酮, 继续反应4 h得到21,收率68%。
Scheme22
Wang等[29]将1-溴-5-己炔-2-酮及其延长碳链的类似物分别与Ⅳ溶于DMF中,于室温反应3 d得到6-位取代的2-氨基-4-氧代吡咯并[2,3-d]嘧啶类衍生物(26, Scheme 23),收率51%~74%。此方法操作简单,收率较高,并且对反应温度要求较低。
Scheme23
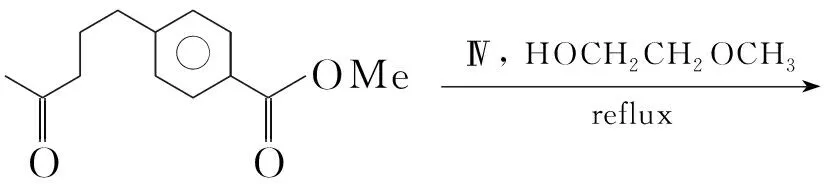
Scheme24
2.2 以Ⅳ的衍生物为原料
Gangjee等[30]将Ⅳ加入2-甲氧基乙醇中,加热至沸腾后加入4-(4-戊酮基)苯甲酸甲酯得到中间体2,6-二氨基-4-氧代嘧啶衍生物(H),收率85%。将H加入二苯醚中,回流反应5 h得到{[2-(2-氨基-6-甲基-4-氧代吡咯并[2,3-d]嘧啶-5-]乙基}苯甲酸甲酯(27, Scheme 24),收率47%。此方法条件温和,但产率较低。
3 结束语
由于2,4-二氨基吡咯并[2,3-d]嘧啶或2-氨基-4-氧代吡咯并[2,3-d]嘧啶类化合物可特异性地作用于DNA合成的两种关键酶,特别是2-氨基-4-氧代吡咯并[2,3-d]嘧啶类化合物还具有二氢叶酸还原酶和胸苷酸合成酶双效抑制剂的作用,因此在抗癌药物研究与开发中具有广阔的发展前景。本研究组曾将2-甲基-4(3H)-喹唑啉酮作为母体,合成了具有氨基二硫代甲酸酯侧链的4(3H)-喹唑啉酮衍生物,其中多个化合物对几种人肿瘤细胞表现出显著的细胞毒活性[31~33]。鉴于吡咯并[2,3-d]嘧啶类化合物是4(3H)-喹唑啉酮类化合物的生物电子等排体,并且具有二氢叶酸还原酶、胸苷酸合成酶双抑制剂的作用,目前本研究组正以2,4-二氨基吡咯并嘧啶和2-氨基-4-氧代吡咯并嘧啶为母体,设计合成其氨基二硫代甲酸酯衍生物,进而研究抗肿瘤活性,以期发现具有多作用靶点的抗肿瘤先导化合物。
[1] Itoh F, Russello O, Akimoto H,etal. Novel pyrrolo[2,3-d]pyrimidine antifolate TNP-351:Cytotoxic effect on methotrexate-resistant CCRF-CEM cells and inhibition of transformylases of de novo purine biosynthesis[J].Cancer Chemotherapy and Pharmacolog,1994,34(4):273-279.
[2] Parenti M D, Pacchioni S, Ferrari A M,etal. Three-dimensional quantitative structure-activity relationship analysis of a set of plasmodium falciparum dihydrofolate reductase inhibitors using a pharmacophore generation approach[J].J Med Chem,2004,47:4258-4267.
[3] Taylor E C, Patel H H, Junt J G. A one-step ring tansformation/ring annulation approach to pyrrolo[2,3-d]pyrimidines.A new synthesis of the potent dihydrofolate reductase inhibitor TNP-351[J].J Org Chem,1995,60:6684-6687.
[4] Gangjee A, Lin X, Sherry F. Design,synthesis,and biological evaluation of 2,4-diamino-5-methyl-6-substituted-pyrrolo[2,3-d]pyrimidines as dihydrofolate reductase inhibitors[J].J Med Chem,2004,47:3689-3692.
[5] Gangjee A, Lin X, Roy L,etal. Synthesis ofN-{4-[(2,4-diamino-5-methyl-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)thio]benzoyl}-L-glutamic acid andN-{4-[(2-amino-4-oxo-5-methyl-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)thio]benzoyl}-L-glutamic acid as dual inhibitors of dihydrofolate reductase and thymidylate synthase and as potential antitumor agents[J].J Med Chem,2005,48:7215-7222.
[6] Gangjee A, Zeng Y, Talreja T J,etal. Design and synthesis of classical and nonclassical 6-arylthio-2,4-diamino-5-ethylpyrrolo[2,3-d]pyrimidines as antifolates[J].J Med Chem,2007,50:3046-3053.
[7] Gangjee A, Hiteshkumar D, Queener S F,etal. The effect of 5-alkyl modification on the biological activity of pyrrolo[2,3-d]pyrimidine containing classical and nonclassical antifolates as inhibitors of dihydrofolate reductase and as antitumor and/or antiopportunistic infection agents[J].J Med Chem,2008,51:4589-4600.
[8] Rosowsky A, Fua H, Queener S F. Synthesis of new 2,4-diamino-7H-pyrrolo[2,3-d]pyrimidines via the taylor ring transformation/ring annulation strategy[J].J Heterocyclic Chem,2001,38:1197-1202.
[9] Rosowsky A, Chen H, Fua H,etal. Synthesis of new 2,4-diaminopyrido[2,3-d]pyrimidine and 2,4-diaminopyrrolo[2,3-d]pyrimidine inhibitors of pneumocystis carinii,toxoplasma gondii,and mycobacterium avium dihydrofolate reductase[J].Bioorg Med Chem,2003,11:59-67.
[10] Gangjee A, Ye Z, Queener S F. Synthesis of three carbon atom bridged 2,4-diaminopyrrolo[2,3-d]pyrimidines as nonclassical dihydrofolate reductase inhibiters[J].J Heterocyclic Chem,2005,42:1127-1133.
[11] Edward C, Taylor, Bin Liu. A new and efficient synthesis of pyrrolo[2,3-d]pyrimidine anticancer agents: alimta(LY231514,MTA),homo-alimta,TNP-351,and some aryl 5-substituted pyrrolo[2,3-d]pyrimidines[J].J Org Chem,2003,68:9938-9947.
[12] Gangjee A, Mavandadi F, Queener S F,etal. Novel 2,4-diamino-5-substituted-pyrrolo[2,3-d]pyrimidianes as classical and nonclassical antifolate inhibitors of dihydrofolate reductases[J].J Med Chem,1995,38:2158-2165.
[13] Zhu G J, Liu Z, Xu Y,etal. Synthesis of prrrolo[2,3-d]pyrimidine analogues of the potent antitumor agentN-{4-[3-(2,4-diamino-7H-prrrolo[2,3-d]pyrimidine-5-yl)propyl]benzoyl}-l-glutamic acid(TNP-351)[J].Heterocycles,2008,75:1631-1638.
[14] Gangjee A, Yanga J, Queener S F,etal. Novel non-classical C9-methyl-5-substituted-2,4-diaminopyrrolo-[2,3-d]pyrimidines as potential inhibitors of dihydrofolate reductase and as anti-opportunistic agents[J].Bioorg Med Chem,2006,14:8341-8351.
[15] Gangjee A, Jain H D, Queener S F. Design,synthesis and biological evaluation of 2,4-diamino-6-methyl-5-substitutedpirrolo[2,3-d]pyrimidines as dihrdrofolate reductase inhibitors[J].J Heterocyclic Chem,2005,42:589-594.
[16] Gangjee A, Zaware N, Raghavan S,etal. Single agents with designed combination chemotherapy potential:Synthesis and evaluation of substituted pyrimido[4,5-b]indoles as receptor tyrosine kinase and thymidylate synthase inhibitors and as antitumor agents[J].J Med Chem,2010,53:1563-1578.
[17] Charles J, Barnett, Grubb L M. Synthesis of pyrrolo[2,3-d]pyrimidines via cyclocondensation of b-alkoxy- and b-amino-a-bromoaldehydes[J].Tetrahedron Lett,2000,41:9741-9745.
[18] Gangjee A, Vidwans A, Elzein E,etal. Synthesis,antifolate,and antitumor activities of classical and nonclassical 2-amino-4-oxo-5-substituted-pyrrolo[2,3-d]pyrimidines[J].J Med Chem,2001,44:1993-2003.
[19] Gangjee A, Dubash N P, KisLiuk R L. Synthesis of novel,nonclassical 2-amino-4-oxo-6-(arylthio)ethylpy rrolo[2,3-d]pyrimidines as potential inhibitors of thymidylate synthase[J].J Heterocyclic Chem,2001,38:349-354.
[20] Gangjee A, Yu J, KisLiuk R L. 2-Amino-4-oxe-6-substituted-pyrrolo[2,3-d]pyrimidines as potential inhibitors of thymidylate synthase[J].J Heterocyclic Chem,2002,39:833-840.
[21] Gangjee A, Zeng Y, McGuire J J,etal. Synthesis of classical,three-carbon-bridged 5-substituted furo[2,3-d]pyrimidine and 6-substituted pyrrolo[2,3-d]pyrimidine analogues as antifolates[J].J Med Chem,2004,47:6893-6901.
[22] Gangjee A, Jain H D, Phan J,etal. Synthesis of 2-amino-4-oxo-5-substitutedbenzylthiopyrrolo[2,3-d]py rimidines as potential inhibitors of thymidylate sythase[J].J Heterocyclic Chem,2005,42:165-168.
[23] Gangjee A, Jain H D, Phan J,etal. Dual inhibitors of thymidylate synthase and dihydrofolate reductase as antitumor agents:Design,synthesis,and biological evaluation of classical and nonclassical pyrrolo[2,3-d]pyrimidine antifolates[J].J Med Chem,2006,49:1055-1065.
[24] Gangjee A, Yang J, McGuireb J J,etal. Synthesis and evaluation of a classical 2,4-diamino-5-substitutedfuro[2,3-d]pyrimidine and a 2-amino-4-oxo-6-substitutedpyrrolo[2,3-d]pyrimidine as antifolates[J].Bioorg Med Chem,2006,14:8590-8598.
[25] Min T, Yi B, Zhang P,etal. Novel furoxan NO-donor pemetrexed derivatives:Design,synthesis,and preliminary biological evaluation[J].Med Chem Res,2009,18:495-510.
[26] Gangjee A, Mavandadi F, Kisliuk R L,etal. 2-Amino-4-oxo-5-substituted-pyrrolo[2,3-d]pyrimidines as nonclassical antifolate inhibitors of thymidylate synthase[J].J Med Chem,1996,39:4563-4568.
[27] Gangjee A, Jain H D, Phan J,etal. 2,4-Diamino-5-methyl-6-substituted arylthio-furo[2,3-d]pyrimidines as novel classical and nonclassical antifolates as potential dual thymidylate synthase and dihydrofolate reductase inhibitors[J].Bioorg Med Chem,2010,18(2):953-961.
[28] Tangeda S J, Garlapati A. Synthesis of new pyrrolo[2,3-d]pyrimidine derivatives and evaluation of their activities against human colon cancer cell lines[J].Euro J Med Chem,2010,45(4):1453-1458.
[29] Wang L, Cherian C, Desmoulin S K,etal. Synthesis and antitumor activity of a novel series of 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolate inhibitors of purine biosynthesis with selectivity for high affinity folate receptors and the proton-coupled folate transporter over the reduced folate carrier for cellular entry[J].J Med Chem,2010,53:1306-1318.
[30] Gangjee A, Yu J, Kisliuk R L,etal. Design,synthesis,and biological activities of classicalN-{4-[2-(2-amino-4-ethylpyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl}-L-glutamicacid and its 6-methyl derivative as potential dual inhibitors of thymidylate synthase and dihydrofolate reductase and as potential antitumor agents[J].J Med Chem,2003,46:591-600.
[31] Cao S L, Feng Y P, Jiang Y Y,etal. Synthesis and in vitro antitumor activity of 4(3H)-quinazlinone derivatives with dithiocarbamate side chains[J].Bioorg Med Chem Lett,2005,15(7):1915-1917.
[32] Cao S L, Jiang Y Y, Feng Y P,etal. Synthesis of substituted benzylamino and heterocyclylmethylamino carbodithioate derivatives of 4(3H)-quinazlinone and their cytotoxic activity[J].Arch Pharm Chem Life Sci,2006,339(5):250-254.
[33] Cao S L, Wang Y, Zhu L,etal. Synthesis and cytotoxic activity ofN-[(2-methyl-4(3H)-quinazolinone-6-yl)methyl]dithiocarbamates[J].Eur J Med Chem,2010,45:3850-3857.
