分子筛催化的戊烯骨架异构反应机理
2011-09-29郭玉华陈标华
郭玉华 蒲 敏 陈标华
(1湖州师范学院,湖州 313000)(2北京化工大学化工资源有效利用国家重点实验室,北京100029)
分子筛催化的戊烯骨架异构反应机理
郭玉华*,1蒲 敏2陈标华2
(1湖州师范学院,湖州 313000)(2北京化工大学化工资源有效利用国家重点实验室,北京100029)
在密度泛函理论中的B3LYP/6-31G(d,p)水平上研究了分子筛催化戊烯骨架异构的微观作用机制,分别对各个基元反应进行了内禀反应坐标(IRC)解析。结果表明:戊烯的骨架异构存在2种反应机理:烷氧基中间体机理和类甲基环丙烷中间体机理。而烷氧基中间体机理又包括2个反应途径,1个是甲基迁移,另1个是乙基迁移。因此,整个异构反应存在3个反应途径。甲基迁移机理和乙基迁移机理都含有3个基元步骤,其中速控步骤分别是甲基迁移和乙基迁移,对应的活化能分别为206.17和207.31 kJ·mol-1,二者几乎相等,表明2个反应途径从能量的角度来说是互相竞争的。类甲基环丙烷中间体机理分2步,碳链扭转和甲基的迁移,反应中间体具有高离子性和高能量的物种。反应速控步骤是碳链扭转反应,其活化能是147.93 kJ·mol-1,明显低于甲基迁移和乙基迁移2个反应途径的能垒,意味着类甲基环丙烷中间体的反应途径更容易发生。
戊烯;分子筛;催化机理;密度泛函理论(DFT)
戊烯经骨架异构制取异戊烯是一个重要的石油化工过程,对汽油辛烷值的改善具有重要意义,已成为近年来广泛关注的课题[1-3]。在各种异构化催化剂中,其中具有独特的孔道结构和表面酸性的沸石分子筛成为主要研究方向[4-8]。由于双键的存在,使得分子筛催化的戊烯异构化反应变得更复杂,通常存在5种类型的竞争反应[4-6]:双键异构、骨架异构、裂解、二聚或多聚反应、氢转移。一般来说,这些反应所需要的酸强度按下列顺序减少:裂解≈聚合>骨架异构>双键异构[9]。在低温只有双键异构被观察到,直链戊烯处在热力学平衡,骨架异构发生在较高温度下,是动力学控制的反应。戊烯的骨架异构是一个放热反应,降低温度在热力学上有利,但由于骨架异构反应比双键异构要困难得多,对反应条件要求较苛刻,反应温度通常在340~450℃。因而,了解烯烃的骨架异构发生机理对抑制副反应,提高产物选择性,改善催化剂性能是非常有意义的。
实验研究提出的丁烯骨架异构机理主要有双分子机理和单分子机理[7,10-14]。大量的研究[14-15]指出单分子机理在正丁烯骨架异构化中占主导地位,双分子机理主要产生副产物。而对于正戊烯和更高烯烃在沸石上的骨架异构化则更是普遍认为它们遵循着单分子机理[6,16-18]。对于丁烯骨架异构的单分子机理,Ponec等[19]提出了4种可能性,其中红外研究结果[20]支持了由丁氧基活性中间体发生的-CH3和-H转移的反应途径以及具有质子化甲基环丙烷类中间产物的反应途径。依据丁烯的单分子机理,戊烯骨架异构化也被认为可以通过这2个反应途径进行。尽管13C NMR和IR谱研究发现了烷氧基活性物种的存在[20-24],但实验研究并不能给出具体的基元反应步骤,无法确定微观作用机制。随着计算机的发展,利用量子化学方法计算分子筛的吸附和反应机理方面的研究也正在兴起。
正戊烯异构化过程与正丁烯异构化过程有相似之处,但正戊烯碳链长,异构产物同分异构体多,因此较之C4烯烃异构化又表现出其独特性和复杂性。Demuth[25]等采用VASP软件包,用局域密度近似方法对2-戊烯的骨架异构化反应进行了研究,计算结果指出2-戊烯的骨架异构通过2位戊氧基中间体机理进行。但2-戊烯质子化也可能生成3位戊氧基中间体,而且2-戊烯骨架异构也可能像丁烯一样存在类甲基环丙烷中间体机理,仅考虑一个反应途径往往并不能很好地解释所观测的实验现象。因此,本课题采用密度泛函方法研究了戊烯骨架异构反应机理,从理论上揭示分子筛催化烯烃异构化反应的微观图像,为实验研究提供理论基础。
1 计算模型与方法
由于分子筛的真实结构体系庞大、结构复杂,目前的计算条件尚不能对分子筛单胞直接进行量子化学计算。考虑到分子筛上发生的反应主要是酸性位附近的原子,通常文献[26-31]中均在分子筛孔道表面截取含有Si、Al四面体的3T或者5T分子簇模型来模拟分子筛B酸位,这个简单的近似已经被证明提供了有利的洞察中间体和过渡态性质的手段。小的簇模型上能够进行全优化获得驻点的真实性质,即绝对最小或鞍点,允许采用高级别的计算方法。此外,在小的簇模型上还能完成内禀反应坐标计算验证反应途径。因此,本文采用Plank等[32]提到的3T簇模型来研究分子筛催化的烯烃骨架异构反应机理。该模型由2个SiO4和1个AlO4组成,边界重原子用OH来饱和,模型中相邻的Si和Al原子用桥O键固定起来,从而对Si-O链产生了锚封作用,适合于理论计算,同时又能表述沸石的典型特征。目前,这个模型已经成功的用于烯烃双键异构机理的理论研究[33-34]。
利用密度泛函理论(DFT)[35]在B3LYP/6-31G(d,p)[36-37]水平上全优化了反应物、3T簇、配位复合物、中间体、过渡态和产物。根据振动分析得到的虚频数目对平衡态和过渡态进行了确认,同时计算零点能 (ZPE)。从过渡态构型出发计算内禀反应坐标(IRC)[38-39],确定过渡态相应的反应物和产物。全部计算工作利用Gaussian 03程序[40]完成。
2 结果与讨论
由于先前已对戊烯的双键异构反应进行了详细的研究[41],这里未考虑1-戊烯到2-戊烯转化以及顺、反异构体之间的转化。仅以反式-2-戊烯为例研究戊烯骨架异构机理。催化反应首先涉及到的是反应物分子在催化剂表面的吸附,当2-戊烯与分子筛接触时,烯烃的双键与酸性位质子发生相互作用形成了π配位的吸附复合物。经计算吸附态的戊烯发生骨架异构反应有2种反应机理:烷氧基中间体机理和类甲基环丙烷中间体机理。而烷氧基中间体机理又包括2个反应途径,1个是甲基迁移,另1个是乙基迁移。因此,整个异构反应存在3个反应途径。
2.1 烷氧基中间体机理
2.1.1 甲基迁移的骨架异构
图1给出了甲基迁移机理的各驻点的几何构型,主要结构参数被列在表1。经IRC分析得出甲基迁移机理包括3个基元反应,首先是吸附态的2-戊烯ta-2Pe质子化生成3位戊氧基复合物a-3Alk,接着这个中间体发生甲基转移得到1位异戊氧基复合物ia-iso-Alk,之后异戊氧基分解得到产物2-甲基-1-丁烯的过程。
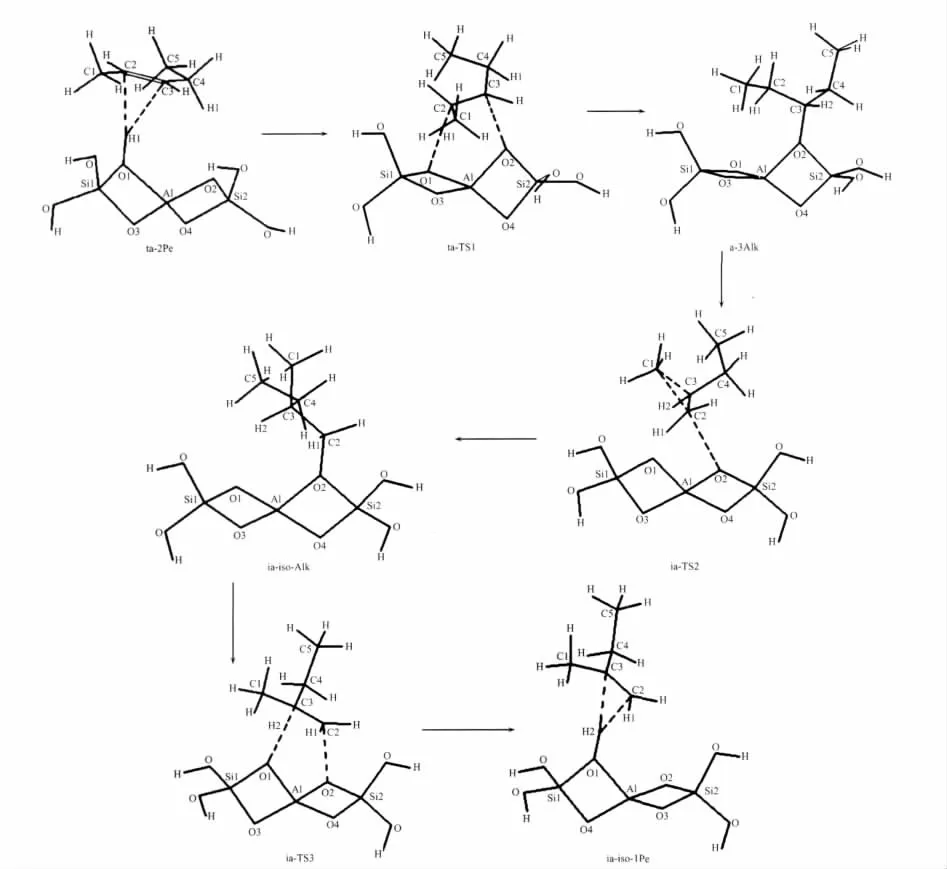
图1 戊烯骨架异构甲基迁移机理各驻点的分子结构Fig.1 Molecular structure of the stationary points for the methyl shift mechanism of the pentene skeleton isomerization
甲基迁移机理关键是3位戊氧基复合物的形成,即ta-2Pe经过渡态ta-TS1到a-3Alk的过程。当2-戊烯吸附在分子筛的酸性位时,沿着反应坐标首先是酸性位质子H1脱离O1而向2-戊烯的C2原子靠近,在过渡态ta-TS1时,C2-H1的距离分别缩短为0.120 3 nm,而H1-O1的距离分别拉长为0.1535 nm,此时C3原子朝着O2原子靠近使得它们的间距分别减小为0.2393 nm。随着反应的进行C2-H1和C3-O2分别缩短为0.1096和0.1483 nm,已达到C-H和C-O共价键的长度,与此同时,碳链也发生较大的扭转,C1-C2-C3和C2-C3-C4角分别减少了9.1°和11.2°,最后生成3位戊氧基中间体(a-3Alk)。此外,C2-C3键也已由双键0.1344 nm转变为单键0.1529 nm。
碳链上甲基迁移是从a-3Alk经过渡态ia-TS2到ia-iso-Alk的过程。随着反应的进行,C1-C2键逐渐拉长直到断裂,各驻点的C1-C2键长分别为0.1533、0.1851和 0.2485 nm; 此时,C1-C3距离逐渐缩短到C-C单键键长,相应的各驻点键长分别为0.2596、0.1812和0.1537 nm,意味着甲基的转移。此外,重要的变化是C3-O2和C2-O2键,从a-3Alk经过渡态ia-TS2到ia-iso-Alk,C3-O2键断裂的同时C2-O2距离缩短为成键的长度,各驻点C3-O2键分别为0.1483、0.2941和0.2469 nm,而C2-O2键分别为0.2440、0.2508和0.1463 nm,预示着甲基迁移的同时烷氧基也由C3原子迁移到C2原子上。2个迁移过程的发生同时引起了C2-C3键在单、双键之间的转变,首先由a-3Alk中的C-C单键到过渡态C=C双键,再到ia-iso-Alk的C-C单键,C2-C3键则分别为0.1529、0.1387和0.1527 nm,键长的变化伴随着C1-C2-C3角的大幅度减少,由115.9°到66.3°最后为35.9°,这些结构参数的变化表明1位甲基基团从C2原子迁移到C3原子的过程,使得正戊基的碳链由直链骨架异构为支链。过渡态ia-TS2构型中包含2个三元环结构,C1C2C3和C2C3O2,这4个原子C1、C2、C3和O2接近在1个平面内,构成的二面角约为165°。三元环结构C1C2C3恰好对应着甲基的迁移,而C2C3O2结构对应着烷氧基的迁移过程。由此可见,整个基元反应键参数的变化是以协同方式进行的。
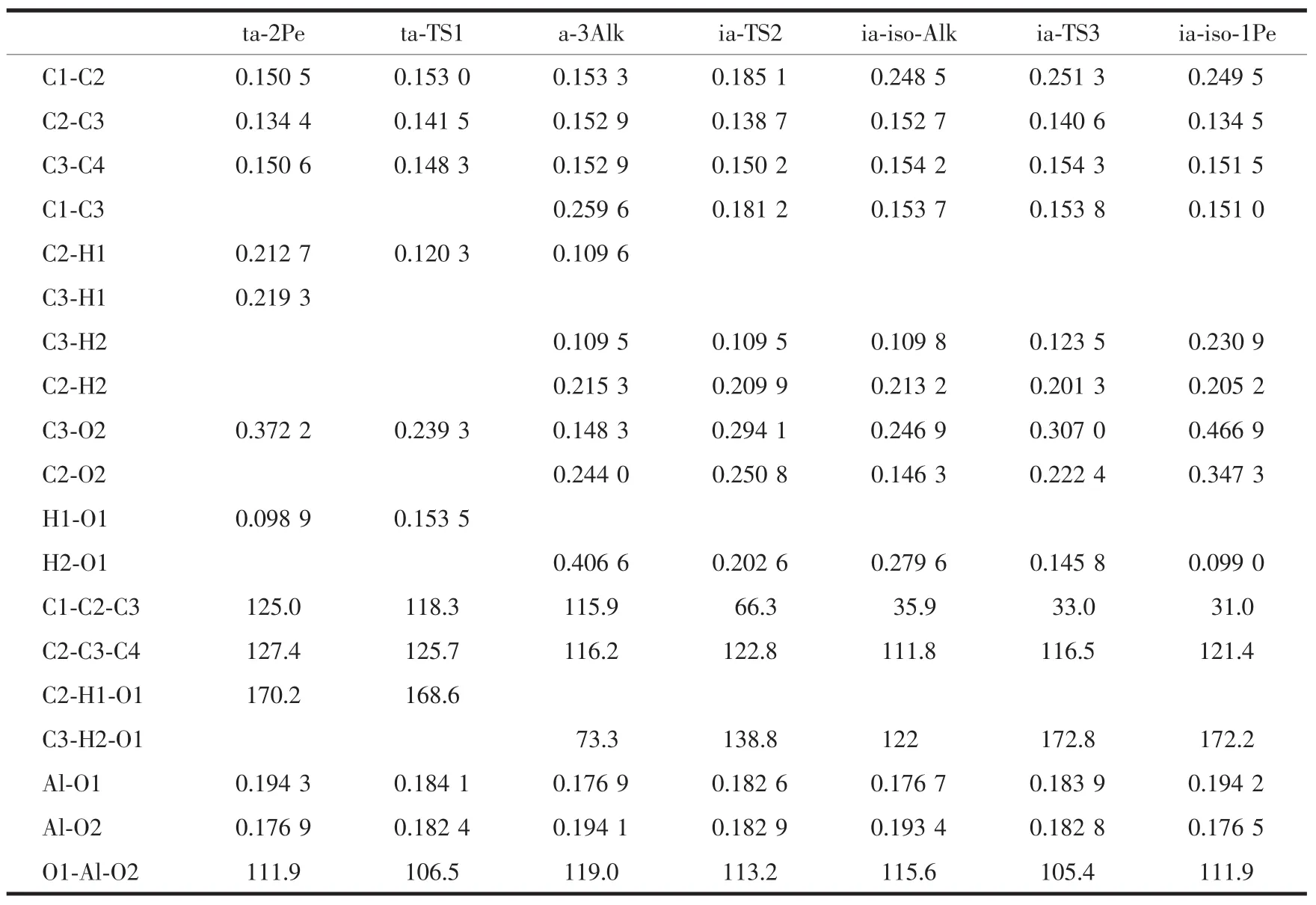
表1 分子筛催化的戊烯骨架异构甲基迁移机理各驻点的几何构型参数Table 1 Bond parameters(distances in nm,angles in°)of the stationary points for the methyl shift mechanism of the pentene skeleton isomerization on zeolites
接下来的基元反应是1位异戊氧基物种分解生成吸附态的1-甲基-2-丁烯的过程,即从ia-iso-Alk经过渡态ia-TS3到ia-iso-1Pe。反应过程中质子的迁移伴随着戊氧基醇盐的分解。沿IRC,H1原子脱离C3原子而向3T簇的O1原子迁移,而C2-O2键长逐渐变长,得到1个六元环的过渡态ia-TS3,此时 C3-H2键拉长为 0.123 5 nm,C2-O2键拉长为0.2224 nm,而H2-O1的距离则由0.2796 nm缩短到0.1458 nm,同时C2-C3键也缩短为0.140 6 nm。之后,C3-H2和C2-O2键断裂,新键H2-O1键形成,C2-C3键由C-C单键转变为C=C双键,生成吸附态的产物复合物ia-iso-1Pe,即1-甲基-2-丁烯π配位复合物。酸性位质子H2与双键两端的碳原子的距离存在一定的差别,与端基的C2原子的距离(0.2052 nm)比与内部的C3原子的距离(0.2309 nm)更近,这是由于2-甲基-1-丁烯中,C3原子上的甲基和乙基的给电子作用,使得电子云偏向C2原子,C2原子带有-0.35 e负电荷,而C3原子带有0.13 e的正电荷,这样C2原子更容易吸取H,从而造成π配位的质子H2偏向C2原子。
2.1.2 乙基迁移的骨架异构
乙基迁移机理不同于甲基迁移,2位戊氧基复合物是这个反应途径的关键中间体。经IRC分析完成乙基迁移也需经过3个基元反应步骤,如图2所示,主要的结构参数被列在表2。首先是2位戊氧基复合物b-2Alk的生成,比甲基迁移多1个产生途径,除了可以由2-戊烯来获得,它还可以通过1-戊烯质子化来生成;之后是这个中间体发生乙基转移得到1位异戊氧基复合物ib-iso-Alk;第三步与甲基迁移机理相同是这个异戊氧基分解产生2-甲基-1-丁烯。

图2 戊烯骨架异构乙基迁移机理各驻点的分子结构Fig.2 Molecular structure of the stationary points for the ethyl shift mechanism of the pentene skeleton isomerization
2位烷氧基中间体的生成,即tb-2Pe经过渡态tb-TS1到b-2Alk的过程。沿着反应坐标,首先是酸性位质子H2质子化2-戊烯的C3原子,使得C3-H2的距离逐渐缩短到成键长度0.1097 nm,同时H2-O1的距离拉长直到断裂;此时C2原子朝着O2原子靠近使得它们的间距缩短为0.1481 nm,形成CO共价键;伴随着键长的变化,C1-C2-C3和C2-C3-C4角也发生较大的扭转,最后形成b-2Alk。
乙基迁移是从b-2Alk经过渡态ib-TS2到ib-iso-Alk的过程。随着反应的进行,C3-C4键由0.1536 nm逐渐拉长直到断裂,此时C2-C4的距离逐渐缩短为0.1547 nm,达到C-C单键键长;而C2-C3键由b-2Alk中的C-C单键(0.1528 nm)转变为过渡态时C=C双键(0.1386 nm),再到ib-iso-Alk的CC单键(0.1528 nm),键长的变化伴随着C2-C3-C4角的大幅度减少由116.5°减少到64.4°最后为35.3°。这些结构参数的变化表明乙基从C3原子迁移到C2原子的过程,使得正戊基由直链骨架异构为异戊基的支链。另外1个重要的变化是C2-O2键由成键长度0.1481 nm逐渐拉长断裂,同时C3-O2距离缩短为共价键的长度0.1462 nm,这说明乙基迁移的同时烷氧基也由C2原子迁移到C3原子上。反应过程结构的变化与甲基迁移机理相似。

表2 分子筛催化的戊烯骨架异构乙基迁移机理各驻点的几何构型参数Table 2 Bond parameters(distances in nm,angles in°)of the stationary points for the ethyl shift mechanism of the pentene skeleton isomerization on zeolites
从ib-iso-Alk经过渡态ib-TS3到ib-iso-1Pe过程是1位异戊氧基物种的分解。与甲基迁移的iaiso-Alk分解相同,沿IRC,C2-H1键断裂的同时新键H1-O1形成,结果H1原子逐渐地远离碳链上的C2原子而靠近3T簇上的O1原子,与此同时,戊氧基复合物ib-iso-Alk上的C3-O2键断裂,戊氧基醇盐发生了分解。另外,整个反应过程伴随着C2-C3键由单键到双键的转变,最后生成了吸附态的异戊烯复合物ib-iso-1Pe。
2.2 类甲基环丙烷中间体机理
图3给出了2-戊烯骨架异构的类甲基环丙烷中间体机理各驻点几何构型,表3列出了主要几何构型参数。IRC分析表明,这个反应机理需要经过2个基元步骤,首先吸附态的2-戊烯复合物ic-2Pe发生碳链扭转生成了类甲基环丙烷中间体ic-Int,之后,ic-Int发生甲基迁移生成了异戊烯的吸附态复合物ic-iso-1Pe,再脱附为异戊烯。图中ic-TS1和ic-TS2分别是碳链扭转和甲基迁移两个基元反应的过渡态。
碳链的扭转反应是指从吸附复合物ic-2Pe经过渡态ic-TS1到中间体ic-Int的过程。在ic-2Pe→ic-TS1过程中,酸性位质子H1质子化双键上的C2原子,使得C2-H1距离从0.2209 nm缩短到0.1103 nm;C3原子上的H2原子吸附到酸性位邻近的O1原子上,而C1原子上的H3原子也朝着活性位O2原子靠近,此时过渡态结构中出现了3个弱的氢键作用,即 H2-O1(0.201 5 nm)、H1-O2(0.216 1 nm)和H3-O2(0.274 5 nm),碳骨架上的 C1-C2和 C2-C3键分别拉长为0.1586和0.1441 nm,而C3-C4键缩短为0.1447 nm,键长变化的同时碳链发生扭转,C1原子处的甲基基团向C3原子扭转,使得C1-C2-C3角由 124.9°变为 108.1°,C1-C3-C2 角由 29.2°变为37.9°,表明碳骨架从平面结构逐渐扭转为立体结构。Mulliken分析表明过渡态具有高的正离子性,具有+0.81e正电荷。从ic-TS1→ic-Int过程,主要的变化是C1原子处的甲基基团继续向C3原子处靠近,使得C1-C2-C3键角由108.1°减到82.3°, 而C1-C3-C2键角由 37.9°增到 53.7°,C1-C2键继续拉长在中间体时已为0.163 9 nm,而C1-C3距离缩短为0.2016 nm。中间体构型中酸性位质子H1与C2原子已经成键,C5H11基团依靠H2-O1(0.1906 nm)、H1-O2(0.228 5 nm)和 H3-O2(0.222 2 nm)这 3个弱的氢键与3T簇活性位发生吸附,此时C5H11基团具有高的正离子性,电荷主要集中在C3原子上。由此看来,可以说这个类甲基环丙烷中间体实际上是1个构型扭转的碳正离子。此外,中间体ic-Int中含有1个C1、C2和C3原子组成的三元环结构,也就是类甲基环丙烷结构。

图3 戊烯骨架异构类甲基环丙烷机理各驻点的分子结构Fig.3 Molecular structure of the stationary points for the methylcyclopropane-like mechanism of the pentene skeleton isomerization
甲基迁移反应是指从中间体ic-Int经过渡态ic-TS2到吸附的异戊烯复合物ic-iso-1Pe过程。这个基元反应中最主要的变化是甲基的迁移和H2原子远离碳链而靠近分子筛的变化。可以看出,过渡态之前主要表现为甲基从C2原子向C3原子的迁移,随着反应的进行C1-C2键迅速拉长在过渡态时已经断裂,其距离为0.2266 nm,此时C1-C3迅速缩短在过渡态时已基本成键,其长度为0.1659 nm,过渡态之后它们的变化减缓。过渡态前C3-H2和H2-O1键长的变化很小,到过渡态时C3-H2键仅拉长了0.0036 nm,而H2-O1距离仅缩短了0.0175 nm,C3-H2距离(0.1139 nm)基本是成键长度,H2-O1距离 (0.1731 nm)还处在断裂状态,因此过渡态之后才主要表现为质子H2从C3原子向分子筛3T簇的O1原子迁移。质子迁移的同时C2-C3键缩短直到C=C双键的形成。整个反应过程同时伴随着弱的氢键H1-O2强度的交替变化,由中间体(0.2285 nm)的弱作用到过渡态(0.2017 nm)的强作用,到产物(0.2993 nm)时这个作用几乎消失,此外,H3-O2之间的作用也逐渐消失。因此可以看出,这个基元反应通过甲基迁移,完成了碳链的骨架异构,生成了异戊烯的吸附态分子;通过质子转移,使分子筛的酸性位得到还原。

表3 分子筛3T簇催化的戊烯骨架异构类甲基环丙烷机理的各驻点的几何构型参数Table 3 Bond parameters(distances in nm,angles in°)of the stationary points for the methylcyclopropane-like mechanism of the pentene skeleton isomerization on 3T cluster of the zeolite
2.3 反应位能面

图4 分子筛催化的2-戊烯骨架异构的能量剖面图Fig.4 Potential energy profile for the mechanism of the skeleton isomerization of 2-pentene on zeolites
图4给出了2-戊烯发生骨架异构的反应能量剖面图,对应的各驻点经零点能校正后的相对能量分别被列在表4。可以看出,烷氧基中间体机理的关键是2位和3位戊氧基物种的形成,即b-2Alk和a-3Alk。生成a-3Alk和b-2Alk的活化能分别为92.94和92.25 kJ·mol-1,这说明生成2种中间体的几率几乎相等。a-3Alk发生甲基迁移的活化能206.17 kJ·mol-1几乎相等于b-2Alk发生乙基迁移的活化能207.31 kJ·mol-1,表明2种中间体的反应能力相近。第三步异戊氧基ia-iso-Alk分解的能垒 (141.18 kJ·mol-1)略高于 ib-iso-Alk分解的能垒 (135.19 kJ·mol-1)。由此从能量的角度来说,2-戊烯发生甲基和乙基迁移的能力几乎相同,反应过程中2个途径是互相竞争的。但由于该反应是在分子筛孔道表面进行的,因此乙基迁移时受到空间位阻的限制要大于甲基迁移。与2-丁烯骨架异构比,2-戊烯的骨架异构多了1个乙基迁移的反应途径,碳链的增长,使得异构反应变得更复杂[21,42]。2个反应机理中位垒最高的基元反应是戊氧基复合物a-3Alk和b-2Alk分别发生的甲基和乙基分子内转移生成ia-iso-Alk和ib-iso-Alk的过程,这说明甲基和乙基迁移过程分别是2个反应途径的速控步骤。
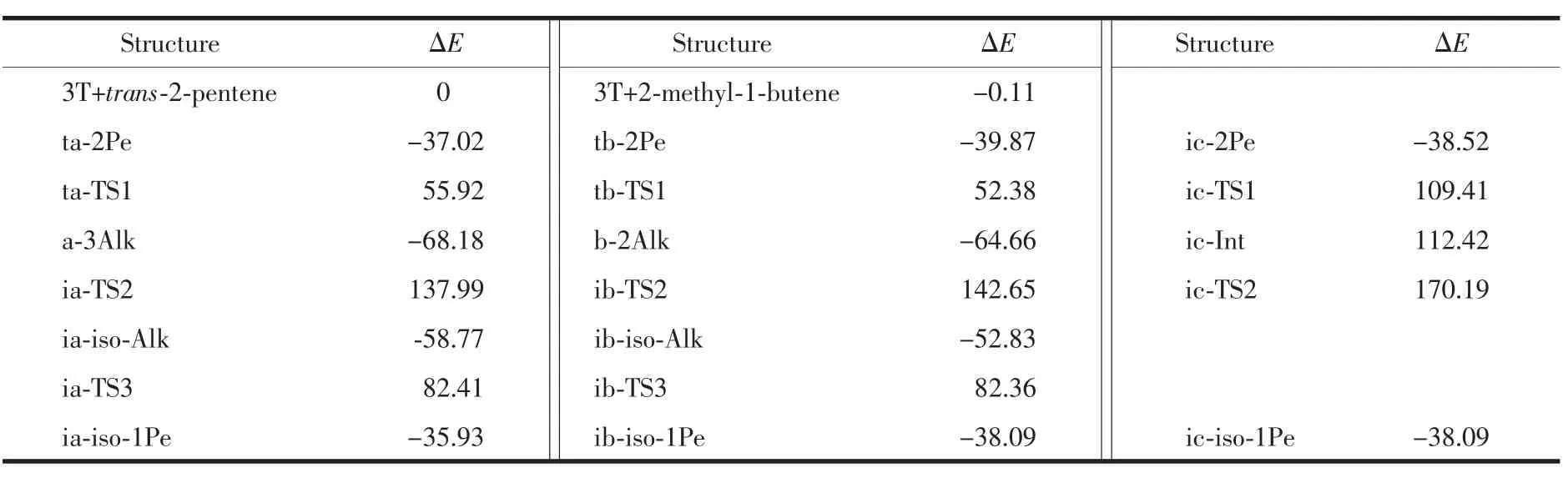
表4 分子筛催化的2-戊烯骨架异构各驻点的相对能量Table 4 Calculated relative energies of the stationary points for the mechanism of the skeleton isomerization of 2-pentene on zeolites(kJ·mol-1)
类甲基环丙烷中间体机理中,ic-2Pe发生碳链扭转的反应位垒是 147.93 kJ·mol-1,从中间体 ic-Int发生甲基迁移的反应位垒是 57.77 kJ·mol-1,这表明碳链扭转的基元反应是速控步骤。中间体ic-Int的相对能量 (112.42 kJ·mol-1)略高于过渡态 ic-TS1(109.41 kJ·mol-1),说明碳链扭转反应中过渡态ic-TS1到中间体ic-Int的势能面非常平缓,意味着中间体是1个高能物种,进一步证明了这个类甲基环丙烷中间体实际上是1个构型扭转的碳正离子。由此可见,对于中间体ic-Int,从热力学角度上说,ic-Int的能量很高;从动力学角度上说,它到过渡态ic-TS1和ic-TS2的位垒都很低。因此中间体ic-Int既是热力学不稳定又是动力学不稳定的,它存在的寿命很短,所以实验很难检测到它,这与烯烃裂解中的碳正离子表现的性质相同[43]。从骨架异构反应速控步骤的能垒看,类甲基环丙烷机理速控步骤的能垒(147.93 kJ·mol-1)要低于烷氧基中间体机理速控步骤的能垒(206.17~207.31 kJ·mol-1),而且类甲基环丙烷中间体发生甲基迁移的能垒(57.77 kJ·mol-1)要远低于前面的烷氧基中间体发生甲基和乙基迁移的能垒,这表明类甲基环丙烷机理也许更容易发生。但由于在分子筛表面烷氧基中间体非常容易形成,因此2个反应机理应该是互相竞争。
综合考虑3个反应途径,以2-戊烯为基准,戊烯骨架异构的表观活化能在138~170 kJ·mol-1范围内。意味着不同的反应条件时,会有不同的反应途径占主导地位。从能量角度来说低温下应有利于低活化能的类甲基环丙烷机理的进行,而高温下2个反应途径也许是互相竞争的。
3 结 论
采用3T簇模型模拟分子筛的酸性位,基于B3LYP/6-31G(d,p)方法,研究了戊烯骨架异构反应机理,依据内禀反应坐标(IRC)理论,分别对各个基元反应进行了IRC解析,得出了戊烯骨架异构的微观作用机制。计算结果表明:
戊烯的骨架异构存在2种反应机理:烷氧基中间体机理和类甲基环丙烷中间体机理。而烷氧基中间体机理包括2个反应途径,1个是甲基迁移,另1个是乙基迁移。二者均包含3个基元步骤,速控步骤分别为甲基迁移和乙基迁移,对应的活化能分别为 206.17 和 207.31 kJ·mol-1。 类甲基环丙烷中间体机理分两步,反应的速控步骤是碳链扭转,其活化能是 147.93 kJ·mol-1。
从反应活化能的角度来说,甲基转移和乙基转移两个反应途径是互相竞争的。由于受到分子筛骨架结构的限制,从空间位阻的角度来说,乙基发生转移反应途径是不利的。类甲基环丙烷中间体机理速控步骤的能垒明显低于烷氧基中间体机理的速控步骤能垒,意味着类甲基环丙烷中间体的反应途径更容易发生。综合考虑3个反应途径,以2-戊烯为基准,戊烯骨架异构的活化能在138~170 kJ·mol-1范围内。低温下类甲基环丙烷机理的占主导地位,而高温下两个反应机理也许是互相竞争的。
[1]Guo J,Cheng X W,Zhou W Z,et al.Microporous Mesoporous Mater.,2005,79:319-328
[2]Sandelin F,Eilos I,Harlin E,et al.Stud.Surf.Sci.Catal.,2004,154:2157-2162
[3]Sandelina F,Salmib T,Murzinb D Y.Chem.Eng.Sci.,2006,61:1157-1166
[4]Hchtl M,Jentys A,Vinek H.Appl.Catal.A,2001,207:397-405
[5]MurerT,Kraushaar-CzarnetzkiB.J.Catal.,1999,187:202-208[6]Fttinger K,Kinger G,Vinek H.Appl.Catal.A,2003,249:205-212
[7]Asensi M A,Corma A,Martínez A.J.Catal.,1996,158:561-569
[8]Tiitta M,Harlin E,Makkonen J,et al.Stud.Surf.Sci.Catal.,2004,154:2323-2330
[9]Corma A,Wojciechowski B W.Catal.Rev.-Sci.Eng.,1982,24:1-65
[10]Houžvicˇka J,Diefenbach O,Ponec V.J.Catal.,1996,164:288-300
[11]Houzvicka J,Ponec V.Catal.Rev-Sci Eng.,1997,39:319-344
[12]Guisnet M,Andy P,Gnep N S,et al.J.Catal.,1996,158:551-560
[13]Szabo J,Perrotey J,Szabo G,et al.J.Mol.Catal.,1991,67:79-90
[14]Houzvicka J,Ponec V.Ind.Eng.Chem.Res.,1997,36:1424-1430
[15]Cˇejka J,Wichterlová B,Sarv P.Appl.Catal.A,1999,179:217-222
[16]Föttinger K,Kinger G,Vinek H.Catal.Lett.,2003,85:117-122
[17]Liu H,Lei G D,Sachtler W M H.Appl.Catal.,1996,137:167-177
[18]Weitkamp J.J.Ind.Eng.Chem.Prod.Res.Dev.,1982,21:550-558
[19]Cheng Z X,Ponec V.Catal.Lett.,1994,27:113-117
[20]Meijers S,Ponec V,Finocchio E,et al.J.Chem.Soc.-Faraday Trans.,1995,91:1861-1869
[21]Haw J F,Richardson B R,Oshiro I S,et al.J.Am.Chem.Soc.,1989,111:2052-2058
[22]Aronson M T,Gorte R J,Farneth W E,et al.J.Am.Chem.Soc.,1989,111:840-846
[23]Lazo N D,Richardson B R,Schettler P D,et al.J.Phys.Chem.,1991,95:9420-9425
[24]Ishikawa H,Yoda E,Kondo J N,et al.J.Phys.Chem.B,1999,103:5681-5686
[25]Demuth T,Rozanska X,Benco L,et al.J.Catal.,2003,214:68-77
[26]Rigby A M,Kramer G J,van Santen R A.J.Catal.,1997,170:1-10
[27]Zheng X,Blowers P.J.Mol.Catal.A:Chem.,2006,246:1-10
[28]Hay P J,Redondo A,Guo Y.Catal.Today,1999,50:517-523
[29]Frash M V,Kazansky V B,Rigby A M,et al.J.Phys.Chem.B,1998,102:2232-2238
[30]Zheng X,Blowers P.J.Mol.Catal.A:Chem.,2005,229:77-85
[31]Rigby A M,Frash M V.J.Mol.Catal.A:Chem.,1997,126:61-72
[32]Plank C J,Drake L C.J.Colloid Sci.,1947,2:399-412
[33]Li H Y,Pu M,Liu K H,et al.Chem.Phys.Lett.,2005,404:384-388
[34]Pu M,Li Z H,Gong Y J,et al.J.Mater.Sci.Lett.,2003,22:955-957
[35]Parr R G,Yang W.Density-functional Theory of Atoms and Molecules.New York:Oxford University Press,1989.
[36]Becke A D.Phys.Rev.A,1988,38:3098-3100
[37]Lee C,Yang W,Parr R G.Phys.Rev.B,1988,37:785-789
[38]Gonzalez C,Schlegel H B.J.Chem.Phys.,1989,90:2154-2161
[39]Gonzalez C,Schlegel H B.J.Phys.Chem.,1990,94:5523-5527
[40]Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian03,Revision B.04,Gaussian,Inc.,Pittsburgh PA,2003.
[41]GuoY H,Pu M,Liu L Y,et al.Comp.Mater.Sci.,2008,42:179-185
[42]Boronat M,Viruela P,Corma A.J.Phys.Chem.A,1998,102:982-989
[43]Stepanov A G,Arzumanov S S,Luzgin M V,et al.J.Catal.,2005,229:243-251
Reaction Mechanism of Pentene Skeletal Isomerization on Zeolites
GUO Yu-Hua*,1PU Min2CHEN Biao-Hua2
(1Huzhou Teachers College,Huzhou,Zhejiang 313000,China)(2State Key Laboratory of Chemical Resource Engineering,Beijing University of Chemical Technology,Beijing 100029,China)
The microcosmic interaction mechanism of pentene skeletal isomerization on zeolites was studied by the density functional theory at the B3LYP/6-31G(d,p)level.The reaction trajectories were determined by the intrinsic reaction coordinate (IRC)methods.The results indicate that the skeletal isomerization of pentene can proceed by two kinds of mechanisms:the alkoxide intermediate mechanism and methylcyclopropane-like intermediate mechanism.The alkoxide intermediate mechanism involves two reaction pathways:methyl shift and ethyl shift.Accordingly,the overall skeletal isomerization of pentene has three reaction pathways.Both the methyl and ethyl shift mechanisms consist of three elementary steps.The rate determining steps are the shift of the methyl group and the shift of the ethyl group,respectively.The corresponding activation barriers are nearly equivalent(206.17 and 207.31 kJ·mol-1,respectively),indicating that two reaction pathways compete between each other.The methylcyclopropane-like intermediate mechanism includes two elementary steps:the torsion of the carbon chain and the methyl shift.This intermediate has highly ionic character and is a high energy species.The rate determining step is the torsion of the carbon chain,and its activation barrier is 147.93 kJ·mol-1.This value obviously is lower than those of the methyl and ethyl shift process,implying that the methylcyclopropanelike intermediate pathway occurs more easily.
pentene;zeolite;catalytic mechanism;density fuctional theory
O613.72;O614.3+1
:A
:1001-4861(2011)02-0333-10
2010-07-17。收修改稿日期:2010-10-24。
国家重点基础研究发展规划项目(973)(No.2004CB217804)和浙江省教育厅项目(No.ZC200805553)资助。
*通讯联系人。 E-mail:guoyuhua@hutc.zj.cn,Tel:0572-2321166
