高效液相色谱-质谱串联法测定人血浆特拉唑嗪的浓度
2011-07-28侯允天郝光涛郑专杰梁海霞马蒙朱力军刘泽源
侯允天,郝光涛,郑专杰,梁海霞,马蒙,朱力军,刘泽源
(1.解放军总医院老年心血管病研究所,北京 100853;2.军事医学科学院附属医院临床药理室,北京 100071;3.首都医科大学附属北京安定医院药剂科,100088;4.蚌埠医学院药学系,安徽蚌埠 233030)
盐酸特拉唑嗪(Terazosin)是一种新型高效的选择性α1受体阻断药,对动脉和静脉的α1受体有较高的选择性阻断作用,对去甲肾上腺能神经末梢突触前膜α2无明显作用,因此在拮抗去甲肾上腺素和肾上腺素的升压作用时,无促进神经末梢释放去甲肾上腺素及明显加快心率的作用,它是哌唑嗪的类似物,与哌唑嗪比较,特拉唑嗪的降压作用出现较慢,且呈良好的线性量效关系而持续时间不变。但作用时间较长,半衰期(t1/2)较哌唑嗪长2~3倍,每天一次给药即能有效控制24 h血压[1-6],是目前老年高血压并发前列腺肥大患者临床应用的一种较好的降血压药。人体前列腺组织中的受体主要以α1肾上腺素能受体为主,而前列腺平滑肌的收缩主要受α1肾上腺素能受体的调控。因此,特拉唑嗪作为α1受体阻断药能减少前列腺尿道阻力,缓解排尿困难[7-10],是一种治疗良性前列腺增生较好的药物。笔者在本研究建立高效液相色谱-质谱串联(HPLC-MS/MS)法测定人体血浆中特拉唑嗪浓度,为特拉唑嗪药动学探讨提供方法。
1 仪器与试药
1.1 仪器 岛津(SHIMADZU)高效液相色谱-质谱串联联用仪,其中岛津(SHIMADZU)高效液相色谱系统包括二元输液泵、自动进样器、柱温箱;串联质谱仪为API 3200型三重四极杆串联质谱仪,含电喷雾离子化源以及串联三重四极杆质谱检测器。色谱工作站:Analyst 1.4.2数据处理软件(美国 AB公司)。GL-88B旋涡混合仪(海门市其林贝尔仪器制造有限公司);AB104电子分析天平(瑞士 METTLER TOLEDO公司);B600A型低速自动平衡离心机(中国白洋离心机厂);Milli-Q Plus纯水器(美国密理博中国有限公司)。
1.2 试药 盐酸特拉唑嗪对照品(中国药品生物制品检定所提供,含量 99.3%,批号:100375-200502),盐酸哌唑嗪对照品(内标,中国药品生物制品检定所提供,批号:100164-200402),甲醇、乙腈均为色谱纯(DIMA公司),试验用水为去离子水(本实验室自制),其他试剂为AR试剂。
2 方法与结果
2.1 色谱条件 色谱柱:Inertsil-C18柱(2.1mm×150mm,3.0 μm);预柱:Shim-pack GVP-ODS(5mm ×2.0mm);流动相:甲醇-水(2mmol·L-1醋酸铵)=90∶10;流速:0.2mL·min-1;柱温:20℃;进样量:5 μL。
2.2 质谱条件 离子源为ESI源;正离子方式检测;多反应监测(multiple reaction monitoring,MRM)方式;喷射电压:5 500 V;离子源温度:450℃;源内气体1[氮气(N2)]压强:306 kPa;源内气体2(N2)压强:374 kPa;气帘气体(N2)压强:68 kPa;碰撞能量:45 V;DP电压:65 V;碰撞气(N2)压力:40.8 kPa;用于定量分析的离子反应对分别为质荷比(m/z)388.4μm/z 247.4(特拉唑嗪)和 m/z384.2 μm/z 231.3(哌唑嗪,内标)。
2.3 溶液的配制
2.3.1 盐酸特拉唑嗪标准溶液的配制 精密称取盐酸特拉唑嗪对照品11.9 mg,相当于特拉唑嗪10 mg,置于50mL棕色量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得200.0 μg·mL-1特拉唑嗪的标准储备液,取该储备液1.25mL置于50mL量瓶中,加入甲醇稀释至刻度,摇匀,即得5 000 ng·mL-1特拉唑嗪甲醇溶液;再以甲醇依次倍比稀释,配成浓度分别为250,100,25,10,5,2 和 1 ng·mL-1的特拉唑嗪标准溶液。
2.3.2 内标(哌唑嗪)标准溶液的配制 精密称取盐酸哌唑嗪对照品11.0 mg,置于10mL棕色量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得100.0 μg·mL-1的内标储备液,取该储备液加入甲醇倍比稀释至100.0 ng·mL-1,作为内标工作溶液待用。对照品溶液于4℃冰箱保存待用。
2.4 样品预处理 精密量取血浆样品200 μL,置10mL磨口玻璃试管中,分别加入100 ng·mL-1哌唑嗪甲醇溶液(内标溶液)40 μL、甲醇 40 μL、饱和碳酸钠溶液100 μL,漩涡混合30 s,再加入叔丁基甲醚2mL,充分涡旋震荡2 min,2 500 r·min-1离心5 min,取上清液叔丁基甲醚置于旋口玻璃试管中40℃水浴氮气挥干,残渣用200 μL流动相复溶,进样5 μL进行HPLCMS/MS分析。
2.5 分析方法确证
2.5.1 质谱分析 取特拉唑嗪和哌唑嗪标准储备液适量用甲醇稀释成浓度为1.0 μg·mL-1的甲醇溶液,按“2.2”项操作,相应的二级全扫描质谱图见图1。
2.5.2 相对基质效应考察 分别取6个不同受试者的空白血浆200 μL,置于10mL磨口玻璃试管中,按“2.4”项下操作,进样量为5 μL,每个受试者空白血浆各做3份;并同时做一条随行的标准曲线,根据标准曲线计算血浆样品实测浓度。结果显示所有试验测定值与标示量的相对标准偏差(RSD)均在±20% 之间,精密度为9.07%(<15%),则可认为没有基质效应干扰[11]。
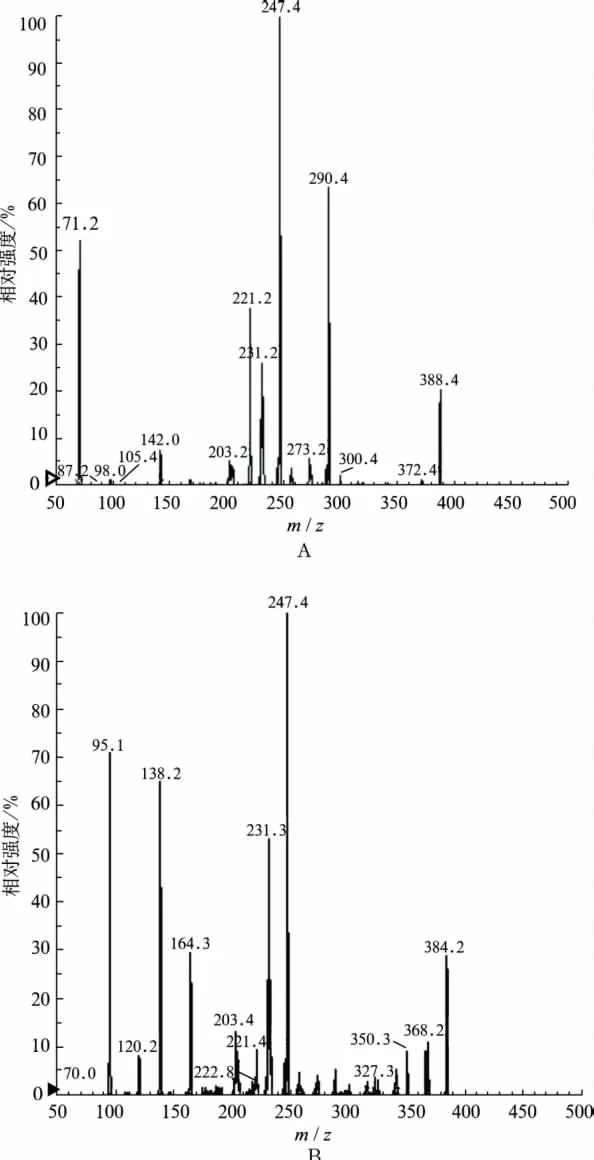
图1 特拉唑嗪和内标哌唑嗪的产物离子扫描质谱图A.特拉唑嗪;B.哌唑嗪(内标)Fig.1 Full-scan production spectra of terazosin and internal standardA.terazosin;B.prazosin(internal standard)
2.5.3 方法专属性 分别取6名受试者的空白血浆200 μL,除不加内标溶液并另外补加甲醇40 μL外,其余按“2.4”项下方法操作,进样5 μL,获得空白样品的色谱图(图2A);将一定浓度的特拉唑嗪标准溶液和内标哌唑嗪溶液加入空白血浆中,依同法进行样品处理,获得相应的色谱图:最低定量限(图2B),标准曲线中间浓度(图2C);取受试者给药后收集的血浆样品,依同法进行样品处理,获得相应的色谱图(图2D)。结果表明,空白血浆中的内源性物质不干扰特拉唑嗪和哌唑嗪的测定。
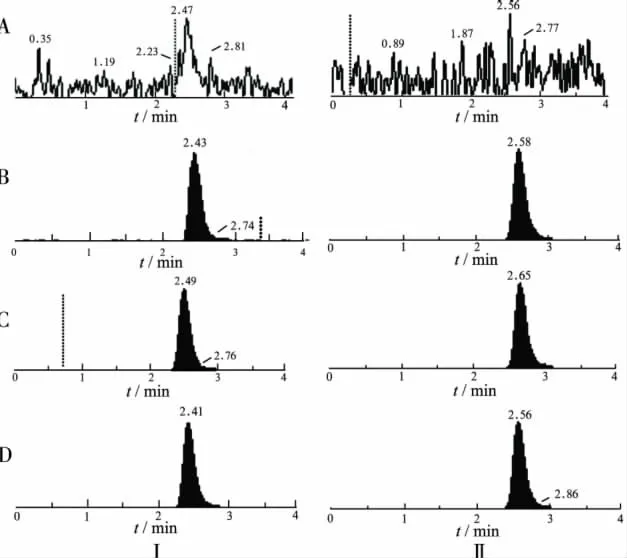
图2 血浆中特拉唑嗪(Ⅰ)和内标哌唑嗪(Ⅱ)的典型MRM色谱图A.空白血浆;B.空白血浆中加入0.2 ng·mL-1特拉唑嗪和100 ng·mL-1哌唑嗪;C.空白血浆中加入2 ng·mL-1特拉唑嗪和100 ng·mL-1哌唑嗪;D.1号受试者第Ⅰ周期第3个点的血浆样品Fig.2 Typical MRM chromatograms of terazosin(Ⅰ)and prazosin(Ⅱ)in plasma samplesA.Blank plasma;B.Blank plasma spiked with 0.2 ng·mL-1 terazosin and 100 ng·mL-1prazosin;C.Blank plasma spiked with 2 ng·mL-1terazosin and 100 ng·mL-1prazosin;D.Plasma sample from No.1 volunteer at the 3rd time point in the 1st periodicity
2.5.4 线性关系考察 取空白血浆200 μL,分别精密加入标准系列溶液的特拉唑嗪对照品溶液和内标溶液各40 μL,涡旋混匀,配成特拉唑嗪浓度分别为0.2,0.5,1,2,5,20 和 50 ng·mL-1的标准含药血浆,按“2.4”项下操作,记录色谱图,以血浆中待测物浓度(X)为横坐标,待测物与内标的峰面积比值(Y)为纵坐标,用加权(w=1/x2)最小二乘法进行回归运算[12],得回归方程:Y=0.006X+0.025 4,r=0.997 3,特拉唑嗪的线性范围是0.2 ~50.0 ng·mL-1,定量下限为0.2 ng·mL-1。
2.5.5 精密度和准确度 按“2.5.3”项分别制备特拉唑嗪低、中、高 3个浓度血浆样品 0.5,2,20 ng·mL-1的质量控制样品,每个浓度6份样品,连续测定3 d,得血浆中特拉唑嗪血浆中低、中、高浓度的平均日内RSD 分别为8.64%,3.77%和 10.55%,日间 RSD 分别为8.15%,9.37%和 9.30%,均<15%,平均相对回收率分别为(100.67±9.03)%,(98.83±4.37)% 和(99.25±11.03)%,低、中、高绝对回收率在分别为75.40%,70.28%,67.39%。
2.5.6 样品稳定性考察 制备血浆样品的低、中、高(0.5,2,20 ng·mL-1)三个浓度的质控样品,分别进行了进样器放置6 h、室温放置4 h、反复冻融3次(-20℃)和长期稳定性(-20℃冰冻放置30 d)4种稳定性的考察,结果显示所有稳定性实验中实测值与添加值的RSD均<15%,表明特拉唑嗪血浆样品在进样器中放置6 h、室温放置4 h、反复冻融3次(-20℃)及-20℃冻存1个月条件下稳定性良好。
3 讨论
笔者在本试验采用HPLC-MS/MS法测定特拉唑嗪血药浓度,具有选择性好、灵敏度高、操作简单等优点。特拉唑嗪在人体血浆中浓度较低,本法测定的定量下限较其他方法低(0.2 ng·mL-1)[13-14]可以更准确测定血浆中的浓度,且方法简便可靠。
特拉唑嗪为生物碱,常制成盐酸盐,故需加入碳酸钠进行中和以使特拉唑嗪游离在有机相叔丁基甲醚中,从而进行液相提取。在氮气流下吹干浓缩可以大大提高分析方法的灵敏度,使样品富集更彻底,不但简化样品处理方法,更减轻受试者的负担,成本较低,更适合于本研究,可见此方法是一种优先的前处理方案。
本试验建立的血浆特拉唑嗪的 HPLC-MS/MS测定法简单,灵敏,适合特拉唑嗪药动学的研究。
[1]丁学琨,潘虹,陈丹丹,等.反向高效液相色谱法测定人血浆中特拉唑嗪片的血药浓度[J].实用药物与临床,2006,9(3):175-176.
[2]孙兴昌,樊朝美,李一石,等.国产多沙唑嗪及特拉唑嗪对比治疗原发性高血压[J].中国新药杂志,2001,10(9):692- 695.
[3]梁高永,徐风,殷献宾,等.特拉唑嗪治疗老年高血压疗效观察[J].临床中老年保健,2002,5(4):272-273.
[4]LYNNE M I,JOHN C C,GARY R,et al.[3H]R-terazosin binds selectively to a1-adrenoceptors over a2-adrenoceptors– comparison with racemic[3H]terazosin and [3H]prazosin[J].Eur J Pharmacol,1997,327(1):79-86.
[5]张志涛,李见春,蒋志文.盐酸特拉唑嗪分散片的人体生物利用度[J].中国医院药学杂志,2009,29(14):1170-1172.
[6]葛乐乐,范辉.特拉唑嗪单芯渗透泵片研制及其控制释放的因素[J].中国医院药学杂志,2007,27(6):740-743.
[7]王荣荣,江振洲,习保民,等.苯丙胺类新化合物抗前列腺增生作用研究[J].中国临床药理学与治疗学,2009,14(7):726-731.
[8]吴声滨,钟巧青,杨天伦,等.多沙唑嗪治疗轻中度高血压的疗效及对血浆内皮素和降钙素基因相关肽的影响[J].医药导报,2005,24(3):189-192.
[9]王珍珊,李玲,邓晓兰,等.液质联用(LC-MS)法检测特拉唑嗪及应用[J].中国临床药理学与治疗学,2010,15(5):530-534.
[10]YOSHINORI T,NAOYA M,NAOKI I,et al.Urodynamic effects of terazosin treatment for Japanese patients with symptomatic benign prostatic hyperplasia[J].J Urol,2002,167(6):2492-2495.
[11]MATUSZEWSKI B K,CONSTANZER M L,CHAVEZENG C M.Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS[J].Anal Chem,2003,75(13):3019-3030.
[12]钟大放.以加权最小二乘法建立生物分析标准曲线的若干问题[J].药物分析杂志,1996,16(5):343.
[13]丁翔宇,蒋晔,安普丽,等.HPLC法测定人血浆中盐酸特拉唑嗪的浓度[J].中国药房,2007,18(14):1076-1078.
[14]PHAIK Y C,KAH H Y,MEN L L.Improved high-performance liquid chromatographic analysis of terazosin in human plasma[J].J Chromatography B,2000,745(2):439-443.
