Kupffer cells contribute to concanavalin A-induced hepatic injury through a Th1 but not Th17 type response-dependent pathway in mice
2011-07-03
Hangzhou, China
Kupffer cells contribute to concanavalin A-induced hepatic injury through a Th1 but not Th17 type response-dependent pathway in mice
Lin Chen, Xiao-Jun Xie, Yu-Fu Ye, Lin Zhou, Hai-Yang Xie, Qin-Fen Xie, Jiong Tian and Shu-Sen Zheng
Hangzhou, China
BACKGROUND: Increasing evidence suggests that a close interaction of Kupffer cells with T cells plays a central role in concanavalin A-induced hepatic injury in mice, but the underlying mechanisms remain obscure. The present study aimed to determine the relative roles of Th1 and Th17 type responses in concanavalin A-induced hepatic injury in mice, and to investigate whether or not Kupffer cells contribute to hepatic injury via a Th1 or Th17 type response-dependent pathway.
METHODS: Immune-mediated hepatic injury was induced in C57BL/6 mice by intravenous injection of concanavalin A. Kupffer cells were inactivated by pretreatment with gadolinium chloride 24 hours before the concanavalin A injection. The interferon-gamma (IFN-γ) and interleukin-17 (IL-17) pathways were blocked by specific neutralizing antibodies. Hepatic injury was assessed using serum transferase activity and pathological analysis. Expression of inflammatory cytokines within the liver was detected by real-time polymerase chain reaction and immunohistochemistry.
RESULTS: Neutralization of IFN-γ significantly attenuated concanavalin A-induced hepatic injury. However, neutralization of IL-17 failed to suppress the injury. Inactivation of Kupffer cells by gadolinium chloride pretreatment protected against concanavalin A-induced injury and significantly reduced hepatic cytokine levels including TNF-α, IL-6 and IFN-γ but not IL-17.
CONCLUSION: Our findings suggest that Kupffer cells contribute to concanavalin A-induced hepatic injury via a Th1 type response-dependent pathway and production of inflammatory cytokines including TNF-α, IL-6 and IFN-γ.
(Hepatobiliary Pancreat Dis Int 2011; 10: 171-178)
Kupffer cells; interferon-gamma; interleukin-17; concanavalin A; hepatic injury; hepatitis
Introduction
Liver disease is a leading cause of morbidity and mortality worldwide. Irrespective of the cause (such as hepatitis virus infection, toxic insult, autoimmunity, drugs or ischemia/reperfusion), hepatic injury seems to be facilitated by similar immune effector mechanisms common to the various liver diseases.[1]Improved understanding of immune-mediated hepatic injury may help to develop therapeutic approaches to this widespread clinical problem. Much progress has been made over the past years in the understanding of the mechanisms underlying immune-mediated hepatic injury through the use of a well-established murine model of hepatitis induced by concanavalin A (Con A) administration.
The experimental immune-mediated hepatic injury model induced by Con A administration is characterized by a remarkable activation of T cells. Activated Kupffer cells (KCs), CD4+T cells and natural killer T cells are regarded as the most prominent effector cells in this model.[2]Given that depletion of CD4+T cells protects mice from Con A-induced hepatic injury, it is generally accepted that CD4+T cells play an important role in the pathogenesis of this disease.[3]Several linesof evidence have suggested that Th1 cells, secreting interferon-gamma (IFN-γ), contribute to Con A-induced hepatic injury.[4,5]Over the past few years, Th17 cells (a novel subset of CD4+T cells), have been increasingly identified as an important regulator of the inflammatory response. Accumulating experimental work has provided evidence for the importance of interleukin-17 (IL-17, signature cytokine of Th17 cells) in the development and progression of many inflammatory diseases,[6]previously thought to be driven by Th1-mediated inflammation. Initially, IL-17 was defined as a proinflammatory cytokine produced by activated CD4+T cells (Th17 cells). However, more recently, IL-17 has also been shown to be able to suppress inflammatory responses and therefore protect against inflammationinduced injury in several diseases such as atherosclerosis, colitis, and asthma.[7-9]The role of IL-17 and the Th17 type response in mediating hepatic inflammation also remains controversial. In murine immune-mediated hepatitis models, the Th17 type response has been shown to cause either a destructive (proinflammatory) or a protective effect.[10,11]
Recently, activated KCs have been shown to play a central role in the initiation and development of Con A-induced hepatic injury, because the specific blockade of KCs by gadolinium chloride (GdCl3) or clodronate liposomes completely inhibits this disease.[12]The potential mechanisms of KCs in this process include production of a variety of inflammatory cytokines and interaction with other immune cells. Notably, it has been reported that close interactions between KCs and T cells (including natural killer T cells) play a key role in Con A-induced hepatitis in mice.[2]Therefore, we hypothesized that KCs contribute to the pathogenesis of Con A-induced hepatic injury in mice via their abilities to secrete an array of proinflammatory cytokines and to aggravate intrahepatic Th1 and/or Th17 type inflammation. To test this hypothesis, we investigated the relative roles of Th1 and Th17 type responses in Con A-induced hepatic injury using neutralizing antibodies; we further compared the effect of KC inactivation on the hepatic expression of the cytokines IL-6, TNF-α, TGF-β, IL-1β, IFN-γ and IL-17 induced by Con A injection.
Methods
Reagents and animals
Con A and GdCl3were from Sigma-Aldrich (USA) and dissolved in phosphate-buffered saline (PBS) and saline respectively. Anti-F4/80 antibody was from Abcam, Inc. (UK). Anti-mouse IFN-γ and IL-17 monoclonal antibodies (mAbs) from R&D Systems (USA) were used to neutralize mouse IFN-γ and IL-17. Control rat IgG2 was injected at equivalent doses and schedules. Specified pathogen-free 8-10-week-old female C57BL/6 mice were from the Shanghai Experimental Center, Chinese Academy of Sciences (China). The mice were maintained under specific pathogen-free conditions within the animal facility, provided with water and foodad libitum, and housed under a 12-hour light/dark cycle. All animals received care in compliance with the institutional guidelines.
Con A-induced hepatitis
Con A-induced hepatitis was performed as previously described.[4]Mice received an intravenous Con A injection (20 mg/kg body weight) via the caudal vein. To block the KCs, 40 mg/kg GdCl3was injected intravenously 24 hours before Con A administration. Saline was injected as a control. Neutralizing antibodies were injected intraperitoneally in certain groups. Each group contained 6-8 mice. Serum samples were taken and livers were also excised at indicated time points after Con A administration. Alanine transaminase (ALT) and aspartate transaminase (AST) levels were measured using an Automatic Chemical Analyzer (7600-100; Hitachi, Ltd, Tokyo, Japan) in the clinical chemistry laboratory of our hospital.
Histology evaluation and immunohistochemistry
Formalin-fixed paraffin-embedded liver tissues were cut into 4 µm sections, placed on polylysine-coated slides, and stained with hematoxylin and eosin for histological analysis. All specimens were evaluated by a single pathologist unaware of the experimental data and scheme. Slides for immunohistochemistry were processed as described in our previous study.[13]Briefly, tissues were dewaxed and rehydrated followed by antigen retrieval through microwaving in 10 mmol/L sodium citrate buffer (pH 6.0). The sections were blocked with 5% bovine serum albumin for 30 minutes and then incubated with each primary antibody in a humidified chamber at 4 ℃ overnight. After washing in PBS, horseradish peroxidase polymer-linked secondary antibody was added for 60 minutes at room temperature. The sections were then visualized with 3, 3'-diaminobenzidine and counterstained with hematoxylin.
Total RNA isolation and real-time quantitative PCR
Hepatic cytokine mRNA expression levels were determined by real-time quantitative polymerase chain reaction (PCR) as described in our previous study.[13]Total RNA was extracted from each frozen sample using TRIzol reagent (Invitrogen, USA) and then converted to cDNA using M-MLV reverse transcriptase (Promega, Madison, USA) according to the manufacturer's instructions. Real-time quantitative PCR was performed using an ABI PRISM 7500 real-time PCR system and SYBR Green PCR master mix (Applied Biosystems, USA). The gene-specific primers used in PCR analysis are shown in the Table. Relative quantification was performed using the comparative threshold cycles method (2-ΔΔCt) as described in the manufacturer's manual.
Statistical analysis
All tests used were two-tailed. Graphic presentation was performed using the GraphPad Prism®5.0 package. Statistical analysis was performed using SPSS 11.6 for Windows. The appropriate nonparametric test (Mann-Whitney) was used to assess group differences. APvalue less than or equal to 0.05 was considered statistically significant.
Results
Severe hepatic injury after Con A administration in C57BL/6 mice
To confirm hepatic injury after Con A administration, the serum levels of transaminases (ALT and AST) were measured before and at 2, 3, 4, 8, 12 and 16 hours after Con A injection (Fig. 1). The results showed that both ALT and AST levels increased gradually 3 hours after Con A injection, peaked at 12 hours, and weresignificantly higher than those of the control mice. Thus, in most experiments thereafter, transaminase levels were measured at 3 and 12 hours after Con A injection (Fig. 2A). The histological analysis of Con A-challenged livers exhibited multiple areas of portal inflammation, hepatocellular apoptosis and necrosis, which confirmed the injury (Fig. 2B).

Table. Sequences of gene-specific primers used for real-time quantitative PCR
Increased hepatic IFN-γ and IL-17-positive cell infiltration after Con A challenge

Fig. 1. Hepatic injury after Con A administration in C57BL/6 mice. Con A (20 mg/kg body weight) was administered intravenously to C57BL/6 mice. Serum ALT and AST levels were measured at 0, 2, 3, 4, 8, 12 and 16 hours after Con A injection.

Fig. 2. Effect of GdCl3pretreatment on Con A-induced hepatic injury. Hepatic injury was assessed by serum transaminase levels and histological analysis at 3 and 12 hours after Con A injection. *:P<0.05. GdCl3(40 mg/kg body weight) or saline was injected intravenously to C57BL/6 mice 24 hours before Con A administration (B, original magnification ×200).
Hepatic mRNA expression of IFN-γ and IL-17 was measured by real-time PCR before and at 1, 3, 8, 12 and 16 hours after Con A injection. The results showed that the mRNA levels of IFN-γ and IL-17 increased gradually, peaked at 3 hours (20.4-fold and 3.4-fold, respectively), decreased, and returned to the control level at 16 hours (Fig. 3A).
To confirm the upregulated production of IFN-γ and IL-17 in the liver from Con A-challenged mice, immunohistochemistry assays for IFN-γ and IL-17 were performed on paraffin-embedded tissue sections. The results also showed significantly increased infiltration of IFN-γ and IL-17-positive cells, mainly located in the portal area and necroinflammatory region. Few IL-17 and IFN-γ-positive cells were detected in normal liver tissues. These results suggested that both Th1 and Th17 type responses were induced within the liver after Con A injection (Fig. 3B).
Neutralization of IFN-γ but not IL-17 attenuated Con A-induced hepatitis
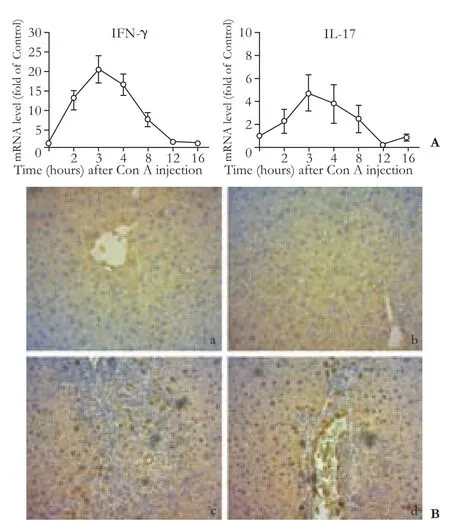
Fig. 3. Increased infiltration of IFN-γ and IL-17-positive cells in liver tissues of Con A-challenged mice. A: Increased hepatic mRNA expression of IFN-γ and IL-17 at different time points after Con A injection. B: representative immunohistochemical staining of hepatic IFN-γ and IL-17 (original magnification ×200). Increased IFN-γ (c) and IL-17-positive cell (d) infiltration were detected in the liver tissues of Con A-challenged mice at 12 hours after Con A injection compared with those of normal control mice (a and b).
We then investigated the relative role of Th1 type and Th17 type responses in Con A-induced hepatic injury by using neutralizing antibodies. Mice were injected intraperitoneally with anti-IFN-γ mAb (0.5 mg/mouse) or anti-IL-17 mAb (0.5 mg/mouse) 2 hours before Con A administration. Serum transaminase levels were measured and livers were harvested for histological analysis at 12 hours after Con A administration. A single injection with the anti-IFN-γ mAb reduced the ALT and AST levels to approximately 12% and 15% of those animals receiving control IgG2α and attenuated the hepatocellular necrosis (P<0.05) (Fig. 4A). However, a single injection with the anti-IL-17 mAb did not reduce, but in fact slightly increased the transaminase levels and aggravated the hepatocellular necrosis compared with the control group, although no significant differences were reached. Histological analysis revealed that massive necrosis was present in the livers of mice treated with control IgG or IL-17 neutralizing antibody, but necrosis was almost absent in animals treated with IFN-γ neutralizing antibody alone (Fig. 4B).
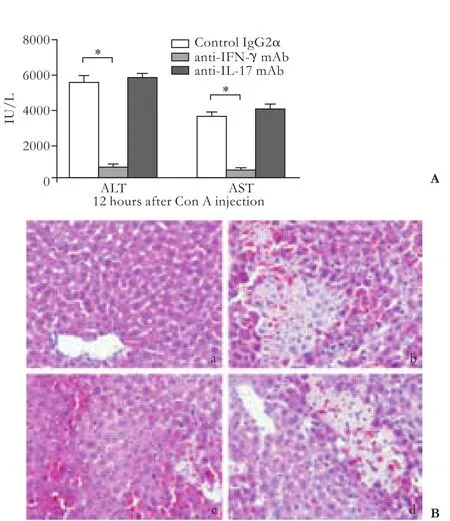
Fig. 4. Effect of IFN-γ or IL-17 neutralization on Con A-induced hepatic injury. Anti-IFN-γ mAb, anti-IL-17 mAb or rat IgG2α were injected intraperitoneally into C57BL/6 mice 2 hours before Con A administration. Hepatic injury was assessed by serum transaminase levels (A, *:P<0.05) and histological analysis (B, original magnification ×200) at 12 hours after Con A injection. (a) normal mice control; (b) control IgG2α; (c) anti-IFN-γ mAb; (d) anti-IL-17 mAb.
KC inactivation by GdCl3pretreatment protected mice from Con A-induced hepatitis
Hepatic F4/80+ KCs greatly increased in the early phase after Con A injection, distributed in the hepatic lobules and periportal area (Fig. 5 A, C). To investigate the mechanism of KCs in the development of Con A-induced hepatitis, KCs were blocked by pretreatment with a single intravenous injection of GdCl3(40 mg/kg body weight) 24 hours before Con A administration. Effective KC inactivation was verified by histological analysis of the cellular colloidal carbon uptake and F4/80 expression in the liver (Fig. 5 B, D). The results showed that F4/80-positive KCs were decreased and colloidal carbon uptake was greatly attenuated by GdCl3pretreatment.

Fig. 5. Effect of GdCl3pretreatment on hepatic KC inactivation by histological analysis of the cellular colloidal carbon uptake and F4/80 expression in the liver. Colloidal carbon (dissolved in saline 1∶50, 1.0 ml/kg body weight) was injected intravenously 20 minutes before blood collection. Liver samples were obtained for further histological analysis (original magnification ×200).
Blockade of KCs by GdCl3significantly inhibited Con A-induced hepatic injury, as indicated by the serum transaminase levels and histological assessment. At 3 and 12 hours after the administration of Con A, the mean ALT levels in GdCl3-pretreated mice were 77.00± 9.82 and 849.67±94.95 IU/L, respectively, significantly lower than those of the saline-pretreated mice at the same time points (266.50±91.35 and 6392.50±1525.60, respectively,P<0.05) (Fig. 2A). In addition, GdCl3pretreatment caused an almost complete abrogation of hepatocellular necrosis and liver inflammation (Fig. 2B).
Effect of KC inactivation on Con A-induced hepatic macrophage-derived cytokine expressionin vivo
Analysis of mRNA expression in liver tissue before and at 1, 3, 8, 12 and 16 hours after Con A administration revealed significantly higher mRNA levels of IL-6, TNF-α, TGF-β, IL-1β, Th1 cytokine IFN-γ and Th17 cytokine IL-17 in Con A-challenged mice compared with those of the controls (Fig. 6A). TNF-α and IL-1β levels peaked within 1 hour, whereas IFN-γ, IL-17, TGF-β and IL-6 levels peaked at 3 hours after Con A injection. Among these cytokines, IFN-γ and TNF-α had the greatest fold increase in expression, 27.10±2.70-fold and 25.08±3.55-fold respectively at the peak point after Con A injection, over the normal liver specimens. ELISA assay confirmed that hepatic IFN-γ and IL-17 levels were increased in Con A-challenged mice compared with those of the controls (Fig. 6B).
Then we examined the effect of GdCl3pretreatment on Con A-induced cytokine mRNA expression in the liver. Pretreatment with GdCl3significantly inhibited the induction of TNF-α, IL-6 and IFN-γ mRNAs expression at the peak point after Con A administration (Fig. 6). However, no significant differences in hepatic mRNA expression of TGF-β, IL-1β and IL-17 were found between the GdCl3and saline pretreatment groups. Furthermore, we measured hepatic IFN-γ and IL-17 protein levels in liver homogenates at 3 hours after Con A injection. The result also showed that IFN-γ secretion was inhibited by GdCl3pretreatment (26.76±1.95 vs. 12.59±1.40 ng/g,P<0.05). However, there was no difference in hepatic IL-17 expression between the GdCl3pretreatment group and control group (6.47±0.76 vs. 5.51±0.86 ng/g,P>0.05).

Fig. 6. Effect of GdCl3pretreatment on hepatic cytokine expression (*:P<0.05). A: Relative hepatic cytokine mRNA levels at the peak after Con A injection were measured by real-time quantitative PCR. B: ELISA measurement of IFN-γ and IL-17 in liver homogenates at 3 hours after Con A injection.
Discussion
Con A-induced hepatitis in mice is commonly used as an experimental animal model for investigating the basic mechanisms of immune-mediated liver injuryin vivo. In the present study, neutralization of IFN-γ by anti-IFN-γ mAb significantly attenuated the hepatic injury induced by an injection of Con A; however, neutralization of IL-17 failed to inhibit the hepatic injury. KC inactivation by GdCl3pretreatment protected against Con A-induced hepatic injury, and inhibited hepatic cytokine secretion including IL-6, TNF-α and Th1 cytokine IFN-γ.
Both IFN-γ and IL-17 have been implicated in inducing and/or aggravating hepatic inflammation and hepatocellular damage in several liver diseases.[13-15]The Th1 type response has been implicated in the pathogenesis of Con A-induced hepatitis;[4]however, the role of the Th17 type response is still controversial. Using the Con A hepatitis model, Nagata found that the signaling of IL-17 and IL-17RA is critical for the development of hepatitis.[10]Lafdil et al[4]reported that deletion of IL-17 slightly reduces Con A-induced hepatic injuryin vivo, and that IL-17 promotes liver inflammation but inhibits hepatocyte apoptosisin vitro. In addition, Zenewicz et al[16]found that IL-17 plays no role in hepatocyte damage during Con A-mediated hepatitis. In the present study, increased infiltration of IFN-γ and IL-17-positive cells was detected in the livers of Con A-challenged mice. Accordingly, we hypothesized that both IFN-γ and IL-17 type responses might contribute to Con A-induced hepatic injury. To test this hypothesis, we used neutralizing antibodies to block the IFN-γ or IL-17 pathway. In accordance with the previous studies describing a key role of the IFN-γ response in mediating Con A-induced hepatitis, neutralization of IFN-γ by pretreatment of mice with anti-IFN-γ mAb significantly reduced serum transaminase levels and attenuated the hepatocellular necrosis induced by Con A. It should be noted that neutralization of IL-17 failed to inhibit Con A-induced hepatitis. These results suggest that Th17 inflammation, although induced within the liver, is not an indispensable requirement for causing hepatic injury induced by Con A injection.
KCs are hepatic macrophages, accounting for 80%-90% of the total tissue macrophages in the body. Although the underlying mechanism remains obscure, KCs have been shown to play a central role in Con A-induced hepatitis by virtue of their ability to secrete a wide variety of cytokines and to act as antigenpresenting cells for initiation and stimulation of local specific immune responses.[12,17]GdCl3, a specific inhibitor of KCs, is often used to study their role. It has been reported that GdCl3pretreatment specifically inhibits the phagocytosis and function of KCs without acting on resident macrophages in other organs.[18]In the present study, KC inactivation by GdCl3pretreatment significantly suppressed the elevation of serum ALT and AST levels, and protected against hepatocellular damage induced by Con A injection, suggesting that KCs are dispensable for Con A-induced hepatic injury. Moreover, we determined the effect of KC inactivation with GdCl3on Con A-induced cytokine expression. We found that KC inactivation significantly inhibited the induction of hepatic TNF-α, IL-6 and IFN-γ expression, and no significant changes in hepatic expression of TGF-β, IL-1β and IL-17 were detected.
Two possibilities can be considered to explain the inhibition of hepatic TNF-α, IL-6 and IFN-γ by GdCl3pretreatment: 1) KCs as the main cellular source of the cytokines upon Con A challenge; and 2) Con A-induced cytokines in the liver as a result of lymphocyte activation. IFN-γ is the predominant Th1 cytokine and has been shown to play an important role in both innate and adaptive immune responses. Evidence that KCs express MHC class II molecules, ICAM-1, as well as low levels of CD80 and CD86, suggests that KCs may act as antigen-presenting cells in the liver. Antigen presentation by KCs has been implicated in regulation of the local and systemic immune responses.[19]In the present study,in vivoblockade of KCs using GdCl3led to reduced hepatic IFN-γ expression and suppression of hepatic injury induced by Con A injection. We therefore speculate that Con A-induced IFN-γ expression in the liver may be a result of T-cell activation, and that the local interactions of KCs with CD4+T cells contribute to an exacerbated intrahepatic Th1 inflammation upon Con A stimulation, although we did not discriminate the cellular source of these cytokines in our study. Our observations are consistent with those of a previous report by Morita et al.[20]It should be noted that KC inactivation did not affect IL-17 mRNA expression in Con A-induced hepatic injury. Although our previous study has shown that KCs have the capacity to induce differentiation of Th17 cells by secreting IL-6 and TGF-β,[21]the results of the present study do not support the hypothesis that KCs aggravate the Con A-induced hepatitis via a Th17 response-dependent pathway.
Activated macrophages are important sources of several cytokines, including IL-6, TNF-α, IL-1β and TGF-β, and they play an important effector role in cell-mediated immune responses. In our study, KC inactivation by GdCl3pretreatment significantly inhibited TNF-α and IL-6 mRNA induction in the liver after Con A administration. Thus, KCs may be the source of TNF-α and IL-6 in the liver. In the presentstudy, TNF-α was the cytokine having the greatest fold increase in expression over the normal liver specimens. Thus we conclude that TNF-α produced by KCs may be the essential effector in Con A-induced hepatitis. Actually, several studies have demonstrated the essential role of TNF-α in the development of Con A-induced hepatitis in mice.[22,23]It is worth mentioning that Nakashima et al[24]reported that blockade of KCs from the liver by GdCl3pretreatment completely inhibited Con A hepatitis, whereas increased serum TNF and IFN-γ levels were not inhibited at all. We believe that Con A-induced cytokine protein level in the plasma does not accurately reflect the Con A-induced T cell activation in the liver.
Considering the findings described above, we propose the following potential mechanism for the development of Con A-induced hepatic injury. After the injection of Con A, activated KCs produce and secrete various proinflammatory cytokines, particularly TNF-α, IL-6 and IFN-γ, and simultaneously activate T cells. The increased production of IFN-γ by KCs and activated lymphocytes results in a predominant Th1 type response within the liver. Activated CD4+T cells and natural killer T cells further produce a large amount of IFN-γ. The augmented production of TNF-α, IL-6, IFN-γ and other inflammatory cytokines within the liver contributes to the development of Con A-induced hepatic injury. Our study provides important insights into the cellular mechanism of Con A-induced hepatic injury. However, there are some limitations. First, we have not provided the direct evidence for an interaction between KCs and T cells (including Th1 cells). Second, intracellular cytokine staining of isolated hepatic lymphocytes needs to be done in future studies.
It has been recognized that a variety of liver diseases of different causes, including alcoholic, autoimmune, drug-induced and viral hepatitis share common immune mechanisms that leads to hepatic inflammation and liver injury. A better understanding of the interaction between KCs and T cells, or involvement of other immune cells in the pathogenesis of this model may help to develop therapeutic approaches to the immune-mediated liver diseases including viral hepatitis, autoimmune hepatitis, drug-induced hepatotoxicity and alcoholic liver diseases in the near future.
Funding: This study was supported by grants from the National Basic Research Program of China (973 Program) (2009CB522403), the Key Program of the National Natural Science Foundation of China (30730085), and the National Natural Science Youth Foundation of China (J20090846).
Ethical approval: Not needed.
Contributors: ZSS proposed the study. CL and YYF wrote the first draft and analyzed the data. All authors contributed to the design and interpretation of the study and to further drafts. ZSS is the guarantor.
Competing interest: No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
1 Herkel J, Schuchmann M, Tiegs G, Lohse AW. Immunemediated liver injury. J Hepatol 2005;42:920-923.
2 Schümann J, Wolf D, Pahl A, Brune K, Papadopoulos T, van Rooijen N, et al. Importance of Kupffer cells for T-celldependent liver injury in mice. Am J Pathol 2000;157:1671-1683.
3 Yamanaka A, Hamano S, Miyazaki Y, Ishii K, Takeda A, Mak TW, et al. Hyperproduction of proinflammatory cytokines by WSX-1-deficient NKT cells in concanavalin A-induced hepatitis. J Immunol 2004;172:3590-3596.
4 Lafdil F, Wang H, Park O, Zhang W, Moritoki Y, Yin S, et al. Myeloid STAT3 inhibits T cell-mediated hepatitis by regulating T helper 1 cytokine and interleukin-17 production. Gastroenterology 2009;137:2125-2135.e1-2.
5 Jaruga B, Hong F, Kim WH, Gao B. IFN-gamma/STAT1 acts as a proinflammatory signal in T cell-mediated hepatitis via induction of multiple chemokines and adhesion molecules: a critical role of IRF-1. Am J Physiol Gastrointest Liver Physiol 2004;287:G1044-1052.
6 Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol 2009;27:485-517.
7 Taleb S, Romain M, Ramkhelawon B, Uyttenhove C, Pasterkamp G, Herbin O, et al. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J Exp Med 2009;206:2067-2077.
8 O'Connor W Jr, Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol 2009;10:603-609.
9 Schnyder-Candrian S, Togbe D, Couillin I, Mercier I, Brombacher F, Quesniaux V, et al. Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med 2006;203: 2715-2725.
10 Nagata T, McKinley L, Peschon JJ, Alcorn JF, Aujla SJ, Kolls JK. Requirement of IL-17RA in Con A induced hepatitis and negative regulation of IL-17 production in mouse T cells. J Immunol 2008;181:7473-7439.
11 Wondimu Z, Santodomingo-Garzon T, Le T, Swain MG. Protective role of interleukin-17 in murine NKT cell-driven acute experimental hepatitis. Am J Pathol 2010;177:2334-2346.
12 Hatano M, Sasaki S, Ohata S, Shiratsuchi Y, Yamazaki T, Nagata K, et al. Effects of Kupffer cell-depletion on Concanavalin A-induced hepatitis. Cell Immunol 2008;251:25-30.
13 Ye Y, Xie X, Yu J, Zhou L, Xie H, Jiang G, et al. Involvement of Th17 and Th1 effector responses in patients with Hepatitis B. J Clin Immunol 2010;30:546-555.
14 Zhang JY, Zhang Z, Lin F, Zou ZS, Xu RN, Jin L, et al. Interleukin-17-producing CD4(+) T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology 2010;51:81-91.
15 Gao B. Cytokines, STATs and liver disease. Cell Mol Immunol 2005;2:92-100.
16 Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 2007;27:647-659.
17 Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol 2009;27:147-163.
18 Neyrinck AM, Cani PD, Dewulf EM, De Backer F, Bindels LB, Delzenne NM. Critical role of Kupffer cells in the management of diet-induced diabetes and obesity. Biochem Biophys Res Commun 2009;385:351-356.
19 You Q, Cheng L, Kedl RM, Ju C. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology 2008;48:978-990.
20 Morita A, Itoh Y, Toyama T, Fujii H, Nishioji K, Kirishima T, et al. Activated Kupffer cells play an important role in intra-hepatic Th1-associated necro-inflammation in Concanavalin A-induced hepatic injury in mice. Hepatol Res 2003;27:143-150.
21 Xie X, Ye Y, Zhou L, Jiang G, Xie H, Feng X, et al. Küpffer cells promote acute rejection via induction of Th17 differentiation in rat liver allografts. Transplant Proc 2010; 42:3784-3792.
22 Morris AM, Sennello JA, Fayad RA, Eckel RH, Dinarello CA, Fantuzzi G. T cell-mediated hepatic inflammation modulates adiponectin levels in mice: role of tumor necrosis factor alpha. Metabolism 2006;55:555-559.
23 Miller AM, Zhang JX. Altered endothelin-1 signaling in production of thromboxane A2 in kupffer cells from bile duct ligated rats. Cell Mol Immunol 2009;6:441-452.
24 Nakashima H, Kinoshita M, Nakashima M, Habu Y, Shono S, Uchida T, et al. Superoxide produced by Kupffer cells is an essential effector in concanavalin A-induced hepatitis in mice. Hepatology 2008;48:1979-1988.
Received December 31, 2010
Accepted after revision February 11, 2011
Author Affiliations: Zhejiang University School of Medicine, Hangzhou 310003, China (Chen L, Xie XJ and Ye YF); Key Laboratory of Combined Multi-organ Transplantation, Ministry of Public Health; Department of Hepatobiliary and Pancreatic Surgery (Zhou L, Xie HY, Xie QF and Zheng SS), Department of Nephrology (Tian J), First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, China
Shu-Sen Zheng, MD, PhD, Key Laboratory of Combined Multi-organ Transplantation, Ministry of Public Health, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, China (Tel: 86-571-87236570; Fax: 86-571-87236466; Email: shusenzheng@zju.edu.cn)
© 2011, Hepatobiliary Pancreat Dis Int. All rights reserved.
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Is the pancreas affected in patients with septic shock?
-- a prospective study - Hepatobiliary & Pancreatic Diseases International (HBPD INT)
- Expression of HLA-G in patients with hepatocellular carcinoma
- GPC3 fused to an alpha epitope of HBsAg acts as an immune target against hepatocellular carcinoma associated with hepatitis B virus
- Outcomes of loco-regional therapy for down-staging of hepatocellular carcinoma prior to liver transplantation
- Cholangiocarcinoma accompanied by desmoid-type fibromatosis