千金藤素对大鼠体内非索非那定药动学的影响
2011-06-01胡玉琴蒋学华于志瀛陶勇黄美兰
胡玉琴,蒋学华,于志瀛,陶勇,黄美兰
(1.广东食品药品职业学院,广州 510520;2.四川大学华西药学院临床药学与药事管理学系,成都 610041)
非索非那定(fexofenadine)是特非那定在人体的活性代谢产物,是组胺H1受体拮抗药[1],用于治疗季节性过敏性鼻炎及慢性特发性荨麻疹。近年来国外研究认为[2-3],非索非那定的吸收与肠黏膜上P-糖蛋白(P-glycoprotein,P-gp)密切相关。P-gp介导的肠道分泌可以降低非索非那定的口服吸收。千金藤素(cepharanthine)是从防己科千金藤属植物的块根中提取分离出的一种双苄基异喹啉类生物碱单体化合物,具有抗炎、抑菌、调节免疫等多种生物学活性[4]。国内文献[5]表明,其有逆转多药耐药的作用,效果明显优于经典的P-gp逆转药,且细胞毒性作用远低于传统的逆转药。为此,笔者研究了千金藤素对非索非那定在大鼠体内药动学的影响,为将千金藤素开发为潜在的P-gp抑制药提供依据。
1 材料
1.1 仪器 LC-10AT高效液相色谱仪[包括SPD-10AV紫外检测器,LC-10AT泵(日本岛津)],Genius 3漩涡混合仪(德国IKA公司),Anke TGL-16C台式离心机(上海安亭科学仪器厂)。
1.2 试药 非索非那定原料药(杭州华东医药集团康润制药有限公司,含量>99%),非索非那定对照品(中国药品生物制品检定所,批号:100852-200601),非那西丁原料药(佳木斯鹿灵制药有限责任公司,含量:>99.5%,批号:0703035),非索非那定混悬液(自制,精密称取30 mg非索非那定原料药,置10 mL量瓶,用20%甘油稀释定容至刻度,摇匀,得每毫升含非索非那定3.086 8 mg的胃灌注液),千金藤素原料药(昆明长春花科技有限公司提供,含量:93%,批号:20060319P),甲醇、丙酮为色谱纯,水为二级纯化水,其他试剂为分析纯。
1.3 动物 雄性SD大鼠[四川大学华西实验动物中心,动物合格证号:0003481,平均体质量为(250±20)g]。
2 方法与结果
2.1 给药方法 SD大鼠10只,雌雄各半,随机分成两组,每组5只,禁食24 h,自由饮水。A组灌胃给予3.086 8 mg·mL-1非索非那定胃灌注液,B组灌胃给予非索非那定浓度为 3.086 8 mg·mL、千金藤素浓度为2.007 mg·mL-1的胃灌注液,均10 mL·kg-1。分别于灌胃后 0,0.083,0.25,0.5,1,1.5,2,3,4,6,8,12 h尾静脉取血约0.35 mL,置0.5 mL肝素化离心管,8 000 r·min-1离心5 min,分离血浆,并于-20 ℃保存。
2.2 血药浓度测定
2.2.1 色谱条件 色谱柱:DikmaTMC18柱(5.6 mm×250 mm,4.6 μm),流动相:乙腈-0.5% 磷酸二氢钾(pH3.8)-三 乙 胺 (30∶ 70∶ 0.3),流 速:1.2 mL·min-1,检测波长:220 nm,柱温:40℃;进样量:20 μL。
2.2.2 标准曲线的绘制 取大鼠空白血浆100 μL,精密加入非索非那定对照品系列浓度甲醇液20 μL,使血浆浓度分别为 0.098,0.196,0.392,0.980,1.960,4.200,7.000 μg·mL-1。再于各管中精密加入14 μg·mL-1非那西丁(内标)溶液 20 μL,充分混合后加入乙腈 300 μL。涡旋 5 min,15 000 r·min-1离心5 min,取上清液置于另一离心管中,50℃下氮气挥干后,用流动相75 μL 溶解,涡旋1 min,取20 μL 进样,记录色谱图与峰面积。以非索非那定与内标峰面积比值(Y)为纵坐标,非索非那定浓度(X)为横坐标,用最小二乘法线性回归,得回归方程:Y=0.590 3X-0.042 2(r=0.999 7)。表明血浆样品在0.098~7.000 μg·mL-1范围内线性关系良好,定量限为0.098 μg·mL-1。
2.2.3 方法专属性考察 在“2.2.1”项条件下,分别将空白血浆、空白血浆加内标和非索非那定、血浆样品,按“2.2.2”项下处理,进样,色谱图见图1。非那西丁的保留时间约10.8 min,非索非那定的保留时间为14.6 min,内源性杂质不干扰内标及非索非那定的分离测定。
2.2.4 方法回收率与精密度实验 按“2.2.2”项下方法制备高、中、低浓度质控样品,连续3 d测定,并与标准曲线同时进行,考察方法回收率及日内、内间精密度(表1)。
2.2.5 血浆样品与相关溶液的稳定性考察 取0.2,5.0 μg·mL-1非索非那定对照品甲醇溶液,室温放置24 h后测定(n=3),药物剩余率分别为(99.73±0.60)%(n=3)和(101.8±2.1)%(n=3)。另将浓度为 0.02,0.5 μg·mL-1的质控样品,置-70 ℃保存 7 d后测定,药物剩余率分别为(99.6±3.6)%(n=3)和(106.4±1.4)%(n=3)。高、低浓度的质控样品按“2.2.2”项下处理后放置12 h测定,药物剩余率分别为(102.2±0.6)%(n=3)和(103.8±6.6)%(n=3)。结果表明,非索非那定在上述存放状态下基本稳定。

图1 空白血浆(A)、空白血浆加非索非那定对照品和内标(B)和大鼠给药后血浆样品(C)的HPLC色谱图1.内标;2.非索非那定Fig.1 HPLC chromatograms blank plasma(A)and blank plasma spiked with fexofenadine and internal standard(I.S)(B)and plasma sample after iv administration of fexofenadine(C)1.I.S;2.fexofenadine

表1 方法回收率和日内、日间精密度实验结果Tab.1 The recovery,inter-and intra-day precision μg·mL-1,n=5
2.4 药动学实验结果 A、B组大鼠给药后血药浓度-时间关系曲线呈二室开放模型(图2),药动学参数见表2。
从图2、表2可以看出,A、B组的血药浓度-时间曲线形状相似,均于1~3 h后达峰,且该曲线存在多峰现象。与A组比较,B组血药浓度升高,AUC0-t、Cmax增大明显,AUC0-t增加 7.09%(P<0.01),Cmax增大15.67%(P<0.05)。tmax、MRT、Ka变化不明显(P>0.05)。表明合用千金藤素后,非索非那定的吸收增加,生物利用度增大。
3 讨论
目前已报道的非索非那定血药浓度测定仅限于高效液相荧光检测法和液质联用法[6-7]。高效液相紫外检测方法一般用来检测制剂配方中非索非那定的含量。而非索非那定是药物转运体P-gp和有机阴离子转运体的底物,由于它在体内不被代谢,常用来作为体内评价P-gp转运行为的探针药物[8-9]。因此,本实验中建立的方法也可用于转运体介导的药动学相互作用的研究。
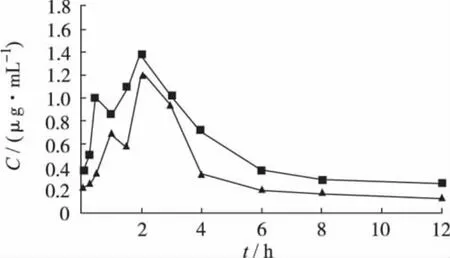
图2 2组灌胃给药后非索非那定药物浓度-时间曲线Fig.2 Plasma concertration-time curves of fexofenadine after oral administration in the 2 groups
表2 2组大鼠非索非那定的主要药动学参数Tab.2 Main pharmacokinetic parameters of fexofenadine the 2 groups ±s
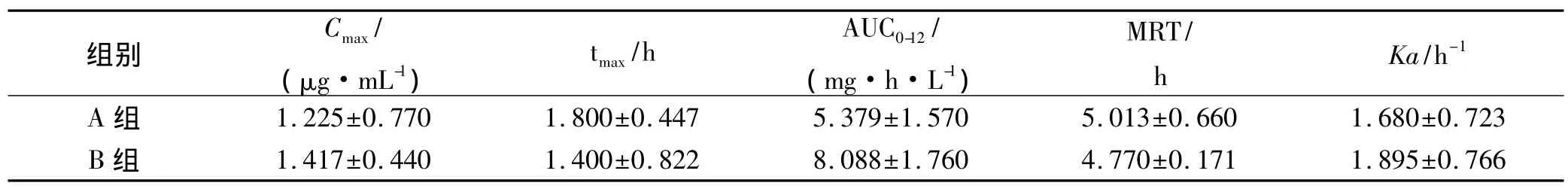
表2 2组大鼠非索非那定的主要药动学参数Tab.2 Main pharmacokinetic parameters of fexofenadine the 2 groups ±s
组别Cmax/(μg·mL-1)tmax/h AUC0-12/(mg·h·L-1)MRT/h Ka/h-1 A 组 1.225±0.770 1.800±0.447 5.379±1.570 5.013±0.660 1.680±0.723 B 组 1.417±0.440 1.400±0.822 8.088±1.760 4.770±0.171 1.895±0.766
大鼠灌胃给予非索非那定后,其体内药动学过程符合二室开放模型,药物浓度-时间曲线存在多个峰现象。合用千金藤素后,非索非那定Cmax、AUC0-t、绝对生物利用度显著增加,但tmax无明显变化,灌胃给药后均于1~3 h后达峰。近年来研究表明[5],千金藤素可部分逆转细胞的多药耐药性,效果明显优于经典的P-gp逆转药,而非索非那定是P-gp的底物[8-9],因此可以初步认为P-gp可能参与了非索非那定大鼠体内药动学过程。合用千金藤素后,千金藤素可能限制了P-gp外排型转运体而使非索非那定的生物利用度增加。
[1] MARKHAM A,WAGSTAFF A.Fexofenadine[J].Drugs,1998,55(2):269.
[2] CHRISTOPHER B,SAMIR G.Grape fruit juice reduces the qral bioavailability of fexofenadine but not desloratadine[J].Clin Pharmocokinet,2002,41(4):311-318.
[3] GEORGE K,DRESSER M D,DAVID G,et al.Fruit juices inhibit organic anion transporting polypeptide-mediated drug uptake to decrease the oral availability of fexofenadine[J].Clin Pharm Ther,2002,71(1):11-20.
[4] 宋玉成,夏微.盐酸千金藤素逆转EAC/ADR细胞多药耐药性的作用及其机制[J].药学学报,2005,40(3):204-207.
[5] 武亚玲,王庆端.盐酸千金藤素逆转K562/ADR细胞的多药耐药性及其与 bcl-2的关系[J].河南肿瘤学杂志,2005,8(2):93-95.
[6] TSUKASA U,NORIO Y.Liquid chromatographic determination of fexofenadine in human plasma with fluorescence detection[J].J Pharm Biomed Anal,2004,35:937-942.
[7] HOFMANN U,SEILER M,DRESCHER S,et al.Determination of fexofenadine in human plasma and urine by liquid chromatography-mass spectrometry[J].J Chromatogr B,2002,766:227-233.
[8] RUSSELL T,STOHZ M,WEIR S.Pharmacokinetics,pharmacodynamicsand tolerance ofsingle-and multiple-dose fexofenadine hydrochloride in healthy male volunteers[J].Clin Pharm Ther,1998,64(6):612-621.
[9] STOHZ M,ARUMUGHAM T,IIPPERT C,et al.Effect of food on the bioavailability of fexofenadine hydrochloride(MDL16455A)[J].Biopharm Drug Dispos,1997,18(7):645-648.
