高效液相色谱法检测羊鸡组织中氟苯达唑、噻苯达唑及其代谢物残留量的研究
2011-05-29张玉洁应永飞林仙军扎依达贾新建
李 丹,张玉洁,汪 霞,应永飞,林仙军,扎依达,贾新建,薛 毅,仲 锋
(1.中国兽医药品监察所,北京 100081;2.浙江省兽药监察所,杭州 310000;3.新疆兽药饲料监察所,乌鲁木齐 830063;4.中国动物疫病预防控制中心,北京 100026)
氟苯达唑、噻苯达唑属于苯并咪唑类驱虫药,对牛、羊的肝片吸虫、大片形吸虫及前后盘吸虫均有良好的杀灭效果,且毒性较小。因此从1960年开始已经广泛应用于畜牧养殖业中[1]。噻苯达唑对大部分胃肠道线虫,尤其是成虫及特定的幼虫有很好的驱虫效果;氟苯达唑属于抗蠕虫药,具有广谱、高效的特点。氟苯达唑和噻苯达唑的广泛使用在一定程度上对人类造成危害,因此必须采取有效的技术手段,对动物性食品中的有关药物残留进行检测,以确保人类对动物性食品消费的安全。本研究旨在建立一种高效液相色谱法,可同时检测羊组织(肌肉、肝脏、肾脏、脂肪)、鸡组织(肌肉、肝脏)中氟苯达唑、噻苯达唑及代谢物2-氨基氟苯达唑、5-羟基噻苯达唑药物的残留。
1 实验部分
1.1 仪器与试剂 Agilent 1100高效液相色谱仪(配紫外检测器),美国安捷伦公司;AE 240型分析天平;Biofuge Stratos高速冷冻离心机,德国贺利氏公司;乙腈(色谱纯)、甲醇(色谱纯),美国Fisher公司;碳酸钠、甲酸、盐酸、氨水、乙酸铵、异辛烷、正丙醇、无水乙醇(分析纯),北京化工厂;氟苯达唑、2-氨基氟苯达唑、噻苯达唑、5-羟基噻苯达唑对照品,德国Adlershof Gmbh公司,含量均≥99.0%;MCX固相萃取柱(60 mg/3 cc),Waters公司。
1.2 对照溶液的配制 氟苯达唑、2-氨基氟苯达唑、噻苯达唑和5-羟基噻苯达唑标准储备液配制(1 mg/mL):称取氟苯达唑、2-氨基氟苯达唑、噻苯达唑和5-羟基噻苯达唑对照品约10 mg,精密称定,于10 mL量瓶中,加甲醇及滴加10%盐酸溶液溶解后,用甲醇稀释至刻度,制成浓度为1 mg/mL的标准储备液,于-20℃以下保存,有效期6个月。
氟苯达唑、2-氨基氟苯达唑、噻苯达唑和5-羟基噻苯达唑标准工作液配制(10 μg/mL):准确量取标准储备液0.1 mL,于10 mL量瓶中,用甲醇稀释成氟苯达唑、2-氨基氟苯达唑、噻苯达唑和5-羟基噻苯达唑10 μg/mL标准工作液。2~8℃保存,有效期1个月。
1.3 色谱条件 流动相 A相为0.02 mol/L乙酸铵溶液,B相为乙腈,梯度洗脱程序如表1所示。
色谱柱 Hypersil C18,250 mm ×4.6 mm(i.d),粒径 5 μm,或相当者;检测波长 306 nm;柱温40 ℃;进样量 20 μL。
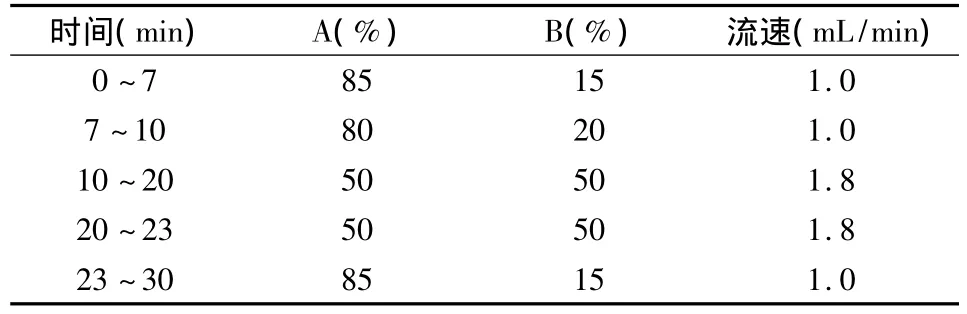
表1 流动相梯度洗脱表
1.4 组织样品的提取与净化 称取(2±0.05)g试料,于50 mL离心管中,加2%碳酸钠溶液0.5 mL、乙腈8 mL、异辛烷5 mL,涡旋混匀,中速振荡5 min,5000 r/min离心5 min,取下层清液于50 mL鸡心瓶中。残渣再加2%碳酸钠溶液0.5 mL、乙腈8 mL重复提取一次,合并两次下层清液,加正丙醇5 mL。于50℃水浴旋转蒸发至干,加入30%盐酸乙醇溶液8 mL,涡旋混匀后备用。
依次用甲醇3 mL、2%甲酸水溶液3 mL,活化MCX固相萃取柱。将备用液过柱。依次用2%甲酸水溶液3 mL、甲醇3 mL淋洗,抽干。用5%氨化甲醇溶液3 mL洗脱。洗脱液于50℃水浴下用氮气吹干。用流动相1.0 mL(0.02 mol/L乙酸铵溶液∶乙腈=85∶15)溶解,混匀。过滤膜后作为试料溶液,供高效液相色谱分析。
2 结果与讨论
2.1 色谱条件的选择 参考国内外有关资料,并对氟苯达唑、2-氨基氟苯达唑、噻苯达唑和5-羟基噻苯达唑进行紫外扫描,考虑要同时检测四种药物,最终确立其检测波长为306 nm。
2.2 标准曲线及线性范围 准确量取适量氟苯达唑、2-氨基氟苯达唑、噻苯达唑、5-羟基噻苯达唑标准工作液,用流动相稀释成浓度分别为0.02、0.04、0.1、0.2、0.3、0.6、1.5 μg/mL 的系列混合标准溶液,将此系列混合标准溶液,依次从低浓度到高浓度按液相色谱条件进行分析,每一浓度进样三次,按其所得三次峰面积的平均值与对应的对照溶液的质量浓度做标准曲线,并计算回归方程及相关系数(表2)。

表2 氟苯达唑、2-氨基氟苯达唑、噻苯达唑和5-羟基噻苯达唑标准曲线
2.3 样品提取 国内外关于测定羊、鸡组织中氟苯达唑、2-氨基氟苯达唑、噻苯达唑和5-羟基噻苯达唑药物残留的报道,有液相色谱-串联质谱、高效液相色谱、荧光检测及紫外检测等方法,但检测波长均有不同。药物残留提取方法的手段也均不相同,有液-液提取、液-固提取等。参考国内外的文献,实验最初采用液 -固提取的方法,使用 C18、HLB、MAX、MCX、硅胶等固相萃取柱,采用的提取液有不同浓度的无机盐溶液及直接用有机溶液提取,经过多次试验反复摸索,最终采用液-固提取,用乙腈作为提取液,从羊、鸡组织中提取药物残留。
在方法研究过程中,分别用甲醇、乙腈、丙酮、乙酸乙酯等有机溶剂作为提取液,再加入少量的酸、碱溶液,以便更有利于药物残留的提取。经过试验摸索用乙腈提取,其回收率较为理想。
在提取实验中,分别在提取液中加入少量的酸、碱溶液。其结果是加入少量的碱溶液比加酸溶液,其效果更为明显。在两次提取时均要加碱,如果仅加一次碱,其回收率明显降低。经过多次试验反复摸索,在用乙腈作为提取液的同时,加入0.5 mL的碳酸钠溶液,提高了提取效率。同时对1%、2%、3%、5%四种不同浓度的碳酸钠溶液进行了考察,实验结果显示,加入2%碳酸钠溶液时提取效率是最高的。因此,最终确立加入碳酸钠溶液的浓度是2%。
在提取脱脂过程中,分别用异辛烷、正己烷等作为脱脂溶剂进行了试验,实验结果显示用异辛烷明显要好于正己烷,因此选用异辛烷作为脱脂溶剂。
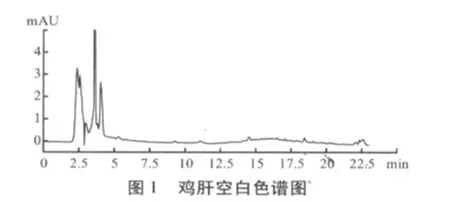
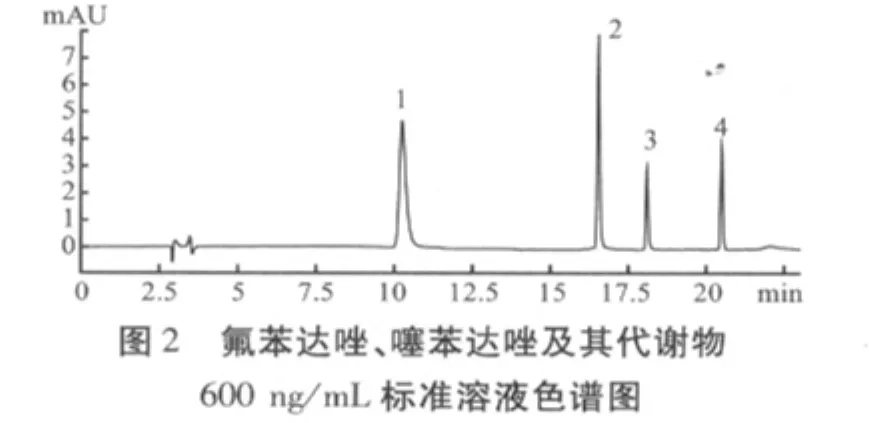
通过上述样品提取方法和色谱条件,空白溶液、标准溶液和试样溶液的高效液相色谱图分别如图1、图2、图3所示。
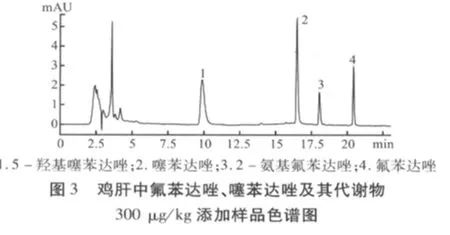
2.4 方法准确度和精密度 采用标准添加法,选取3种不同浓度,药物在羊肝、肾、肌肉、脂肪四种组织的最高残留限量均为100 μg/kg,选取的三种浓度为20、100、200 μg/kg。药物在鸡肌肉中的最高残留限量为 200 μg/kg,选取的三种浓度为 20、200、400 μg/kg。药物在鸡肝脏中的最高残留限量为500 μg/kg,选取的三种浓度为 20、500、800 μg/kg。选取的三种浓度基本包含了上述药物进行残留检测的要求,其中灵敏度为20 μg/kg的浓度点为定量限。每种浓度5个样品,重复3批次,求回收率,批内、批间变异系数。测定结果如表3~表6所示。
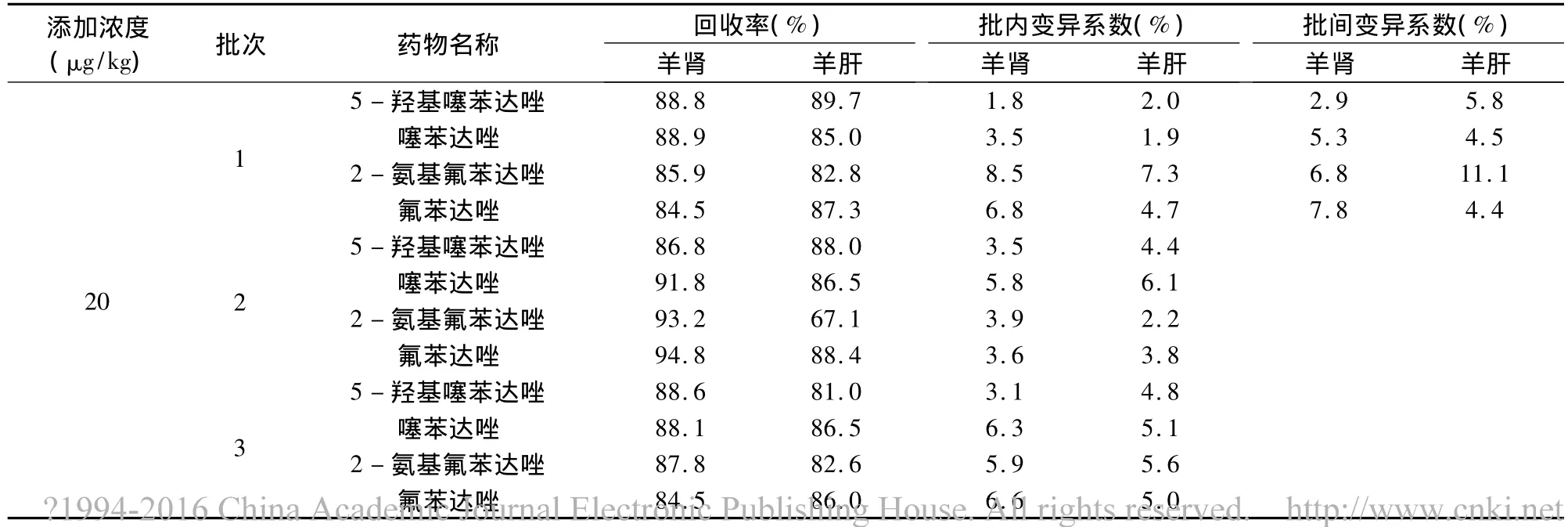
表3 羊肾、羊肝中氟苯达唑、2-氨基氟苯达唑、噻苯达唑和5-羟基噻苯达唑回收率测定结果
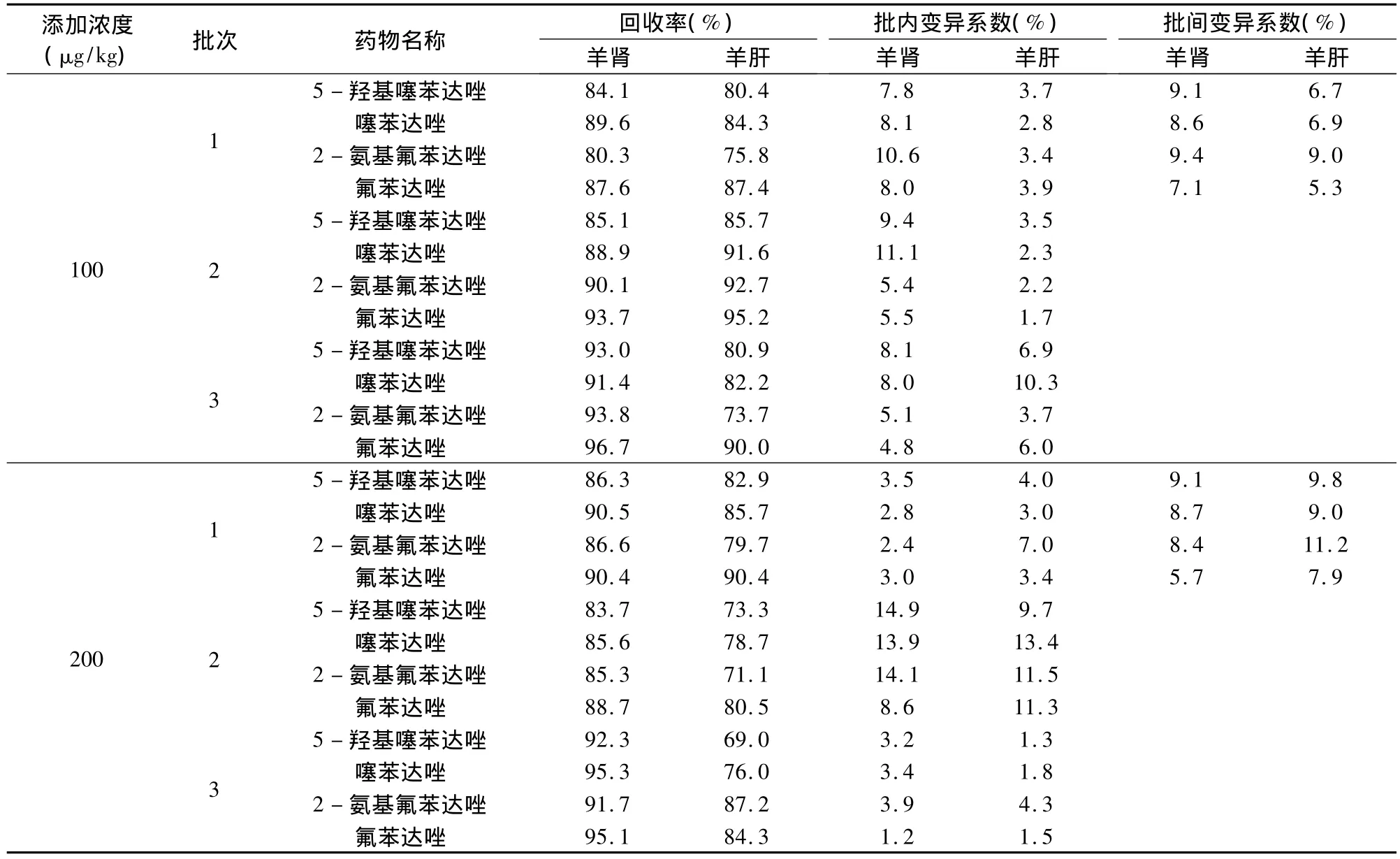
(续表)
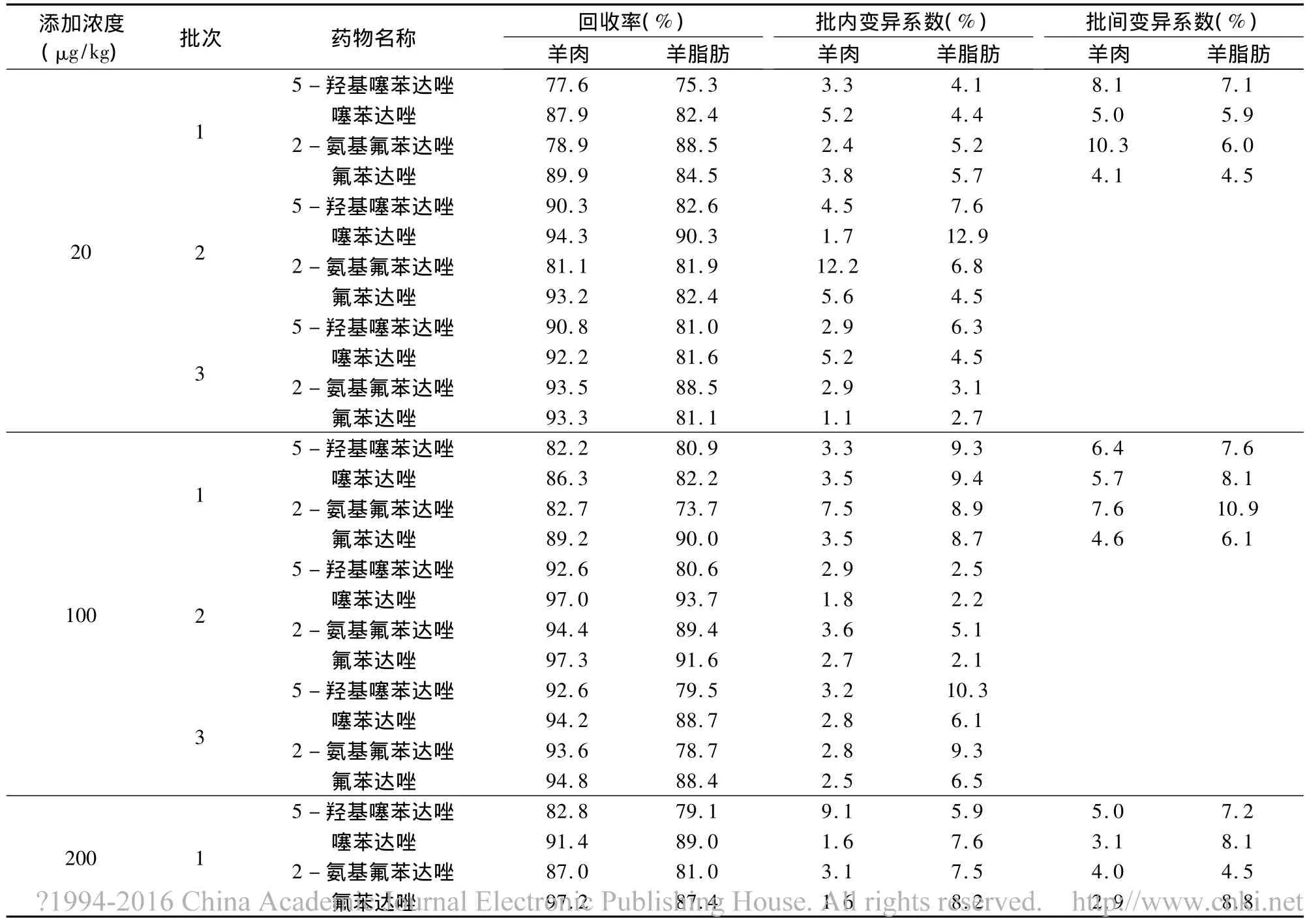
表4 羊肉、羊脂肪中氟苯达唑、2-氨基氟苯达唑、噻苯达唑和5-羟基噻苯达唑回收率测定结果
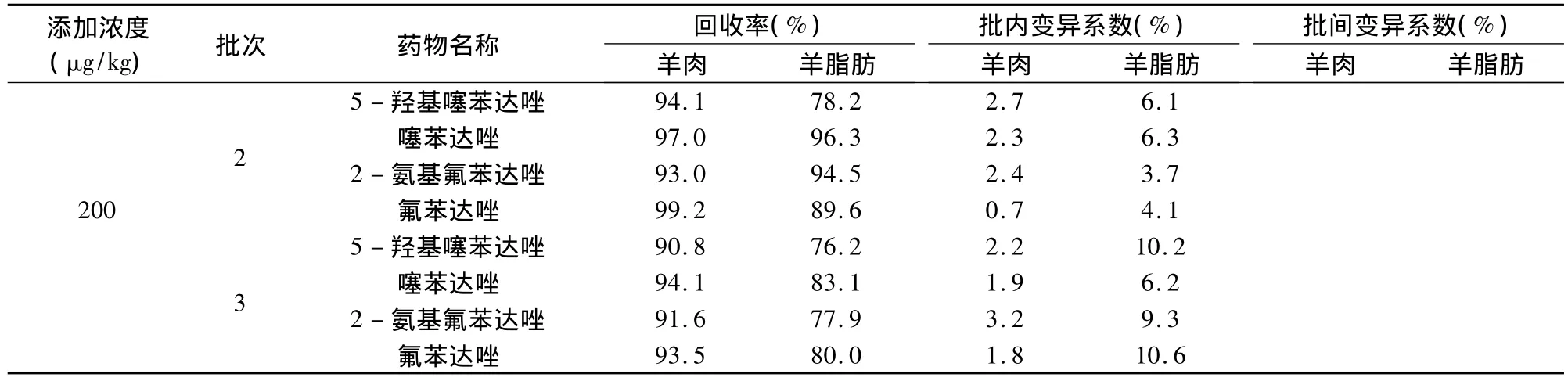
(续表)
从上述表中数据可以看出,无论是各种添加浓度的回收率,还是批内、批间的变异系数均符合农业部农发[2003]1号关于发布《兽药残留试验技术规范(试行)》通知中规定的要求。此方法提取过程简单,操作步骤少,所用化学试剂的量也少,减少了对环境的污染。此方法适用于羊、鸡组织中上述药物残留的检测。

表6 鸡肝中氟苯达唑、2-氨基氟苯达唑、噻苯达唑和5-羟基噻苯达唑回收率测定结果
[1]陈杖榴.兽医药理学[M].第2版.北京:中国农业出版社,2002:282.
