差示扫描量热法对芝麻酚纯度标准物质的定值*
2011-05-26郭永辉周浩辉徐维盛龚宁波
郭永辉 周浩辉 徐维盛 龚宁波 吕 扬
(北京协和医学院,中国医学科学院药物研究所,北京 100050)
差示扫描量热法对芝麻酚纯度标准物质的定值*
郭永辉 周浩辉 徐维盛 龚宁波 吕 扬
(北京协和医学院,中国医学科学院药物研究所,北京 100050)
建立了采用差示扫描量热法对芝麻酚纯度标准物质的定值及不确定度评价的数学模型、有效检测技术和分析方法。采用差示扫描量热法测量芝麻酚样品纯度的实验条件为升温速率3.0 K/min,称样量3.4~4.7 mg,炉内气体为静态空气。对通过均匀性检验和长期稳定性考察的芝麻酚纯度标准物质进行定值和不确定度评价,同时采用高效液相色谱法进行实验结果验证。芝麻酚在3.4~4.7 mg范围内与峰面积呈线性关系(r2=0.999 2,n=6);方法精密度为0.89%(n=6);采用差示扫描量热法及建立的数学模型对芝麻酚纯度标准物质定值及不确定度评价结果为99.46%±0.09%(k=2,P=0.95)。HPLC法测定纯度平均值为99.53%。建立的差示扫描量热法检测技术可实现对芝麻酚纯度标准物质的定值及不确定度评价,该检测分析方法具有简便、快速、准确的特点。
芝麻酚 纯度标准物质 差示扫描量热法 定值及不确定度评价 HPLC
芝麻酚是中药材黑芝麻中的主要有效成分之一,又名3,4-亚甲二氧基苯酚,是芝麻油的重要香气成分和品质稳定剂。芝麻酚具有非常强的抗氧化能力,常用于食品、医药的抗氧化剂,同时它更是合成抗高血压药物、心血管药物的重要起始原料,也是农药胡椒基丁基醚的原料[1,2]。目前芝麻酚在国际上非常紧俏,尤其是药物合成领域的需求量很大。研究芝麻酚纯度标准物质对于我国药物质量控制具有重要的科学意义。
依据我国标准物质研制的技术规范管理要求和定值的一般原则及统计方法[3,4],笔者基于差示扫描量热法(DSC)建立了芝麻酚高纯度标准物质定值检测分析方法和不确定度评价数学模型,为研制国家有证标准物质提供了有效检测技术和分析方法。差示扫描量热法测量物质纯度是依据物质的熔点因杂质成分存在而下降的基本原理。其样品中的杂质成分含量越多,其熔点值下降越大。只有当物质化学纯度中的杂质成分摩尔分数小于2%时,物质熔点的下降与杂质含量的关系可用Van’t Hoff方程表示,即建立不确定度评价数学模型为:

式中:T0——理想中的纯物质熔点值(凝固点),K;
Tm——物质的真实熔点值(凝固点),K;
R——气体常数,即8.314 J/(K·mol);
x——纯物质的摩尔分数;
ΔH——物质的摩尔熔融焓变值,J/mol;
T0-Tm——物质纯度值因各种杂质成分存在而导致的熔点下降量。
目前文献报道芝麻酚纯度测定的方法仅限于高效液相色谱法[5,6],而差示扫描量热法在纯度测定中已经开始广泛应用[7-10]。利用建立的DSC检测芝麻酚纯度分析方法,实现对芝麻酚纯度标准物质的均匀性检验和稳定性考察,完成对芝麻酚纯度标准物质的纯度定值和不确定度评价。利用高效液相色谱法对芝麻酚纯度标准物质的纯度进行了不同原理检测方法的结果验证。结果表明笔者建立的差示扫描量热法测定芝麻酚纯度标准物质的检测分析方法具有简便、快速、准确等优点。
1 实验部分
1.1 主要仪器与试剂
差示扫描量热仪:DSC-1型,标准40 μL 铝坩埚,瑞士Mettler Toledo公司;
电子天平:XS105型,感量0.01 mg,瑞士Mettler Toledo公司;
高效液相色谱仪:1200型,配有DAD检测器,Agilent Chemstation色谱工作站,美国Agilent 公司;
甲醇:HPLC级,美国Thermo Fisher Scientific有限公司;
纯净水:杭州娃哈哈集团有限公司;
一级铟标准物质:GBW13202,熔 点(429.75±0.01) K;
芝麻酚纯度标准物质:中国医学科学院药物研究所。
1.2 实验条件
(1)差示扫描量热法
精密称取芝麻酚纯度标准物质样品适量,置于40 µL的标准铝坩埚中,压盖,放置于差示扫描量热仪中,采用空的40 µL标准铝坩埚作为参比。实验条件:升温速率3.0 K/min,称样量为3.4~4.7 mg,炉内气体为静态空气。记录DSC热流值变化曲线,采用Mettler Toledo公司的STARe分析软件计算其纯度。图1为芝麻酚纯度标准物质DSC检测图谱。

图1 芝麻酚纯度标准物质DSC检测图谱
(2)高效液相色谱法
根据文献报道的高效液相色谱条件[5,6]经优化后进行芝麻酚纯度测定。采用Agilent Eclipse XDB C18(150 mm×4.6 mm, 5 µm)色谱柱;流动相:甲醇-水(40∶60);检测波长:295 nm;流速:1.0 mL/min;柱温:45℃;进样量:10 µL。记录色谱图及峰面积,利用峰面积归一化法进行纯度测定。
2 DSC方法学验证
2.1 线性方程与线性范围
分别精密称取6份3.4~4.7 mg之间不同质量的芝麻酚纯度标准物质,按1.2(1)条件进样测定,记录热流值变化曲线及峰面积。以样品的质量X(mg)为横坐标,吸热峰面积Y为纵坐标绘制标准曲线。试验结果表明,芝麻酚的质量在3.4~4.7 mg内与峰面积呈良好的线性关系,回归方程为Y=115.074 7X+51.518 2,r2=0.999 2(n=6)。
2.2 方法的精密度
分别精密称取6份4.25 mg左右的芝麻酚纯度标准物质,按1.2(1)条件每份测定1次。6份芝麻酚峰面积测定结果的相对标准偏差为0.89%,表明方法的精密度良好。
3 标准物质的均匀性、稳定性验证与定值
3.1 均匀性检验
从500个已经分装成最小包装单元(瓶)中,随机抽取j(15)瓶样品,按1.2(1)条件采用DSC法进行均匀性检验。在重复性条件下对每瓶进行i(3)次独立检测,共获得Xij(45)个数据;采用单因素方差分析对均匀性检验数据进行统计分析。计算每瓶在重复性条件下获得的3个数据的平方和,获得组内平方和数据。计算每一瓶i(3)个数据的平均值,共获得j(15)个平均值,以j个平均值作为一组数据,计算j(15)个数据的平方和,获得组间平方和数据。计算组内和组间各自相应的均方差(MS),并用组间均方差除以组内均方差计算得统计量F值。计算各自的自由度N1(组间)和N2(组内),确定显著性水平α。查F分布表中相应N1、N2、α所对应的F临界值,并与计算获得的统计量F进行比较,若统计量F小于临界值F,则瓶与瓶之间样品非均匀引入的离散性与测量方法引入的离散性相比,可以忽略不计,认为样品均匀;反之样品不均匀。
芝麻酚纯度标准物质均匀性检验结果见表1,方差分析结果见表2。经上述方法分析,得到F值为1.75,查F检验临界值表知:F0.05(14,30)= 2.04,即,F<F0.05(14,30)。结果表明,芝麻酚纯度标准物质的纯度具有良好的均匀性。

表1 芝麻酚纯度标准物质均匀性检验结果

表2 芝麻酚纯度标准物质均匀性检验方差分析结果
3.2 稳定性考察
对均匀性检验合格的样品采用DSC法,在12月内按“先密后疏”的原则对标准物质进行稳定性考察,每个时间点随机取样6瓶,每瓶取样1次,按1.2(1)条件进行检验,以样品纯度为指标考察样品稳定性。按照《国际标准化组织标准物质指南35》[2]规定,以X代表时间,以Y代表标准物质的特性量值(芝麻酚的纯度),拟合成一条直线,获得直线方程为Y= -0.000 002X+0.994 524,则斜率为:

自由度为n-2=6-2=4,P=0.95(95%置信区间)的t因子等于2.7 7 6。由于故斜率是不显著的,因而未观察到不稳定性。因此包装后的芝麻酚纯度标准物质在12月内稳定性良好,稳定性考察数据见表3。

表3 芝麻酚纯度标准物质稳定性考察检测结果
3.3 芝麻酚纯度标准物质DSC法定值
(1)定值测定
随机抽取通过均匀性检验及稳定性考察的芝麻酚纯度标准物质样品10瓶,按1.2(1)条件进样测定,计算其纯度。检测结果见表4。
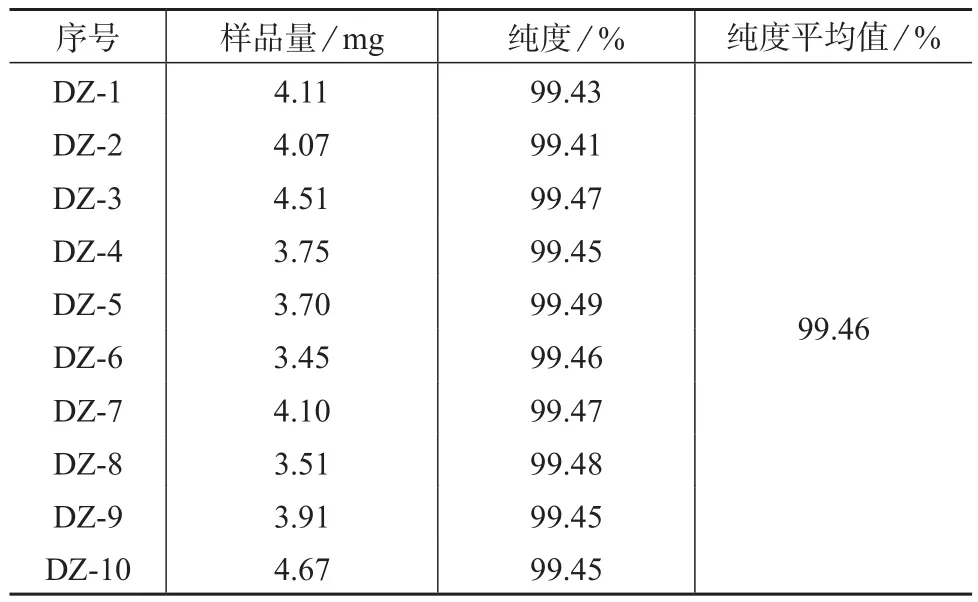
表4 芝麻酚纯度标准物质的DSC纯度检测结果
(2)可疑值剔除—格拉布斯(Grubbs)检验
采用格拉布斯检验对得到的10组芝麻酚纯度标准物质的纯度值进行可疑值剔除,得到的纯度值经计算得G=1.967。查格拉布斯临界值表可知:G0.95(10)=2.176。因G<G0.95(10),故10组数据中无可疑值存在。
(3)标准值确定
通过格拉布斯(Grubbs)检验的10组纯度值的平均值即为标准值。由表4的定值数据确定芝麻酚纯度标准物质纯度的标准值为99.46%。
(4)不确定度评定
标准值的不确定度由3部分组成:(1)通过测量数据的标准偏差、测量次数及所要求的置信水平按统计方法计算;(2)通过对测量影响因素的分析,估计出其大小;(3)物质不均匀性和物质在有效期内的变动性所引起的不确定度。由于该芝麻酚纯度标准物质已通过均匀性检验及长期稳定性考察,因此由物质不均匀性和物质在有效期内的变动性所引起的不确定度可忽略不计。根据JJF 1059-1999[11]及建立的数学模型,不确定度评定结果见表5。

表5 芝麻酚纯度标准物质不确定度评定结果
合成不确定度u为:

扩展不确定度U=ku,k取2,计算扩展不确定度为U=0.000 400 3×2=0.000 800 6,P=0.95。则芝麻酚纯度标准物质纯度定值结果及不确定度表示为:99.46%±0.09%(k=2,P=0.95)。
4 高效液相色谱法方法学验证
4.1 线性方程与线性范围
精密称取芝麻酚纯度标准物质样品24.98 mg,置于25 mL容量瓶中,用流动相溶解并定容至刻度,配制成浓度为999.2 µg/mL储备液。分别精密吸取0.1、0.5、1.0、2.5、5.0、8.0 mL至10 mL容量瓶中,用流动相稀释至刻度,获得10.0、50.0、99.9、249.8、499.6、799.4 µg/mL 6种浓度溶液。按1.2(2)色谱条件进样测定。以芝麻酚浓度X(µg/mL)为横坐标,峰面积Y为纵坐标绘制标准曲线。实验结果表明,芝麻酚纯度标准物质溶液浓度在10.0~799.4 µg/mL内与峰面积呈良好的线性关系,回归方程为:Y=15.871X+56.088,r2=0.999 8(n=6)。
4.2 仪器精密度
精密称取1份12.5 mg的芝麻酚纯度标准物质,置于25 mL容量瓶中,使用流动相定容,按1.2(2)条件进行测定。重复测定6次芝麻酚纯度标准物质峰面积的相对标准偏差为0.16%,表明仪器精密度良好。
4.3 方法精密度
精密称取6份芝麻酚纯度标准物质12.5 mg,置于25 mL容量瓶中,用流动相溶解并定容至刻度,按1.2(2)项下色谱条件进行测定。实验结果表明,测定的6份芝麻酚峰面积的相对标准偏差为0.76%,表明方法精密度良好。
4.4 高效液相色谱法纯度验证
按1.2(2)色谱条件对3份芝麻酚纯度标准物质进行了化学纯度测定。精密称取约12.5 mg芝麻酚纯度标准物质,置于25 mL容量瓶中,用流动相溶解并定容至刻度。每份样品测定3次,利用峰面积归一化法计算纯度值。芝麻酚纯度标准物质高效液相色谱法的定值检测结果见表6。

表6 芝麻酚纯度标准物质的HPLC纯度检测结果
5 结论
采用差示扫描量热法对芝麻酚纯度标准物质进行定值,对定值结果的不确定度进行了评定,并用高效液相色谱法进行结果验证,两种方法测定结果一致。实验结果表明,研制的芝麻酚纯度标准物质为高纯度化学物质。该物质已于2011年5月成功申报为国家一级化学成分纯度标准物质(GBW09538)。
差示扫描量热法具有样品用量少、样品前处理简单、操作快速、不使用溶剂,检测结果准确度高、重现性好等优点。采用差示扫描量热法测定芝麻酚化学纯度的检测方法具有科学性和应用性,为高纯度化学物质的纯度检测提供了一种新的检测技术和分析方法。
鉴于目前用于化学纯度的检测分析方法较为有限,由多种不同原理检测技术对结果验证是分析测量的一种趋势,新技术、新方法的研究和应用将有助于推动科学技术的不断进步和发展。
[1] 国家药典委员会. 中华人民共和国药典一部[M].北京:中国医药科技出版社,2010:323.
[2] 刘雅茹, 冯雪松, 刘俊亭, 等.芝麻酚的合成方法改进[J].解放军药学学报, 2006,22(2):140-142.
[3] JJF 1006-1994 一级标准物质技术规范[S].
[4] CNAS-GL29 标准物质/标准样品定值的一般原则和统计方法[S].
[5] 黄纪念, 宋国辉, 孙强, 等. HPLC测定芝麻油中木脂素类化合物含量研究[J].中国粮油学报, 2011.01:29-32.
[6] 李丹丹, 曾晓雄. 高效液相色谱法测定芝麻油中木酚素的含量[J].湖北农业科学, 2011,50(4):821-824.
[7] 郭永辉, 杨德智, 龚宁波, 等. 采用差示扫描量热技术建立苦参碱纯度测定新分析方法[J].河北医药, 2010,32(24):3 438-3 440.
[8] Mathkar S, Kumar S, Bystol A, et al. The use of differential scanning calorimetry for the purity verification of pharmaceutical reference standards[J]. Journal of Pharmaceutical and Biomedical Analysis,2009, 49: 627-631.
[9] Giron D, Goldbronn C. Place of DSC purity analysis in pharmaceutical development[J]. Journal of Thermal Analysis,1995, 44: 217-251.
[10] 魏莉萍, 林景雪, 马月琴, 等. 差示扫描量热法测量纯度的不确定度分析[J].化学分析计量, 2001, 10(4): 4-6.
[11] JJF 1059-1999 测量不确定度评定与表示[S].
DEFINING VALUE OF SESAMOL PURITY REFERENCE MATERIALS BY DIFFERENTIAL SCANNING CALORIMETRY
Guo Yonghui, Zhou Haohui, Xu Weisheng, Gong Ningbo, Lü Yang
(Peking Union Medical College, Institute of Materia Medica & Chinese Academy of Medical Sciences, Beijing 10050, China)
The analytical method and mathematical model for purity and uncertainty evaluation of sesamol purity reference materials were established. The purity of the substance was measured by the DSC. The experimental conditions were listed below:the heating rate at 3.0 K/min, the sample weight was 3.4-4.7 mg, furnace gas as a static air. The purity and uncertainty value of sesamol was determined, which through the uniformity test and long-term stability inspection. The purity value results were verified by HPLC method. The calibration curves showed good linearity (r2=0.9992,n=6) within tested mass ranges of sesamol (3.4-4.7 mg). The RSD was 0.89%(n=6). The purity value of sesamol purity reference materials determined by DSC was 99.46%±0.09%(k=2,P=0.95), and 99.53% by HPLC. The differential scanning calorimetry method developed in this study was validated to be simple, rapid, accurate, reliable, and was successfully applied to the purity and uncertainty evaluation of sesamol purity reference materials.
sesamol, purity reference materials, DSC, purity value and uncertainty evaluation, HPLC
*科技部科技基础性工作专项重点项目(2007FY130100);重大新药创制“十一五”规划项目(2009ZX09501-021);卫生行业科研专项(200902008-02)
2011-08-17
