新烟碱类化合物几何构型优化方法的选择
2011-03-07王辉宪彭鑫林姜晖霞王玲陶亚
王辉宪,彭鑫林,姜晖霞,王玲,陶亚
(湖南农业大学 理学院,湖南 长沙 410128)
新烟碱类化合物对害虫高效,而对高等动物低毒,符合环保对农药高效低毒的要求,因而受到广泛关注[1]。但新烟碱类化合物的杀虫作用机制研究并不完善,其构效关系的研究还停留在初级阶段[2]。要深入研究新烟碱类杀虫剂与受体的微观作用机制,定量研究化合物电子结构与其活性间的关系,得到准确的化合物电子结构模型是基础。而分子几何构型优化的可靠性是一切精确计算的基本保证[3]。由于理论上对于分子几何构型优化尚无规律和系统方法可循,只能通过一些系统性的比较来探讨。Pople等[4]提出的Gn系列方法、微扰MPn理论能达到很高的精确度,但考虑到研究体系的分子较大,计算量大,费用昂贵,寻找一种计算量适中而结果又相对可靠的计算方法具有实践意义。另外,新烟碱类化合物分子结构较复杂,合成困难,通常缺乏晶体结构数据,它们的分子构型必须选用恰当的量子化学方法进行几何优化,依靠理论预测得到。笔者选用具有晶体结构实测数据的2种新烟碱类化合物分子(化合物1为吡虫啉,化合物2为吡虫啉的N3位甲基取代物)[5]的构型为基点,采用AM1[6]、RHF[7]和B3LYP[8-9]等3种方法,在6-31G、6-31G(d)2种基组水平上进行优化,比较优化所得优势构型与实测的晶体结构数据间的差别,结合考查所需计算精度与时间,选择适合的优化方法,以期为新烟碱类化合物空间构象考察以及量子化学与定量构效关系研究提供依据。
1 试验与方法
1.1 红外光谱扫描与计算
采用溴化钾压片法在QWF-310傅立叶交换红外光谱仪(日本岛津公司)上测定吡虫啉(IMI)标准品(纯度99.2%,购自Sigma公司)的红外光谱。
1.2 分子构型的几何优化方法
利用Gaussview3.07构建2种目标化合物结构。用Hyperchem7.0软件中的MM+方法对化合物进行几何优化,得到初始几何构型;利用Gaussian03软件中的AMl、RHF、B3LYP,借助Berny能量梯度法进一步优化,得到目标分子的5种理论优势构型,并进行频率检查验证。计算机运行环境为Windows XP系统。
2 结果与讨论
2.1 分子振动频率
对2种目标化合物不同方法优化后的振动频率进行计算。结果表明,它们均没有振动虚频,说明所得分子构型均是相对稳定的状态,而不是过渡态[10-11]。
2.2 不同量化方法对键长的影响
在不同的基组水平上,采用AMl,RHF和B3LYP 3种方法优化得到2种目标化合物的键长参数见表1、表2,并与文献值[5]相比较。从整体看来,AM1方法优化所得到的键长参数明显偏离试验值,RHF方法偏离程度次之,而密度泛函方法优化得的结果与试验值最为吻合。用AM1方法优化化合物1的键长平均偏差为0.056 1 Å,其中差别最大在N2–C7键上,其计算值为1.430 8 Å,与试验测量值偏差达0.090 8 Å。许多文献报道,N2原子是吡虫啉重要的活性结构之一[12],所以这种偏差的存在势必给新烟碱类化合物的量子化学研究造成较大影响。化合物2的键长平均偏差略小,但仍有0.038 4 Å。
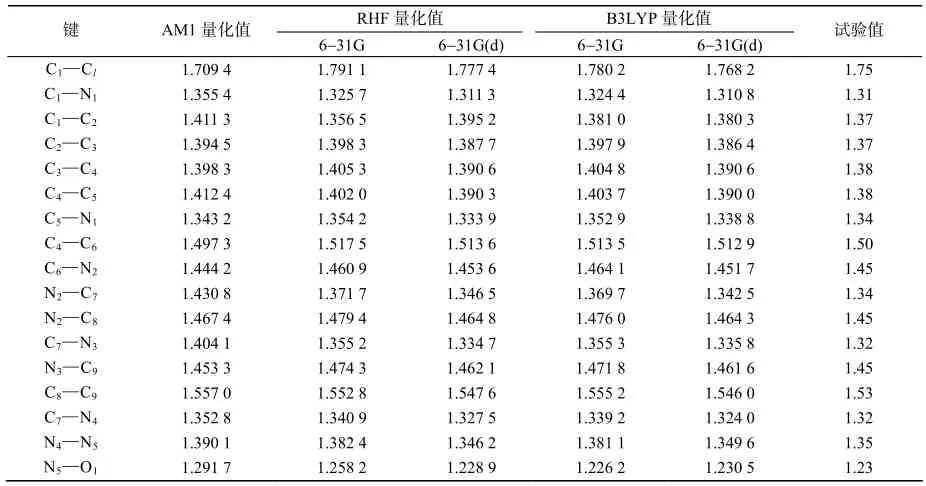
表1 不同方法优化所得的化合物1主要键长的理论值和试验值Table 1 Bond lengths of compound 1 in different methods Å

表2 不同方法优化所得的化合物2主要键长的理论值和试验值Table 2 Bond lengths of compound 2 in different methods Å
在同一计算水平下,不同基组上优化计算的精度不同。相同的RHF方法,选用对外层d轨道极化的基组优化2种目标化合物的键长,平均偏差小于未添加极化函数的基组优化的结果,这种符合程度的提高,在目标化合物2的C1–Cl键上尤为明显,这可能与氯元素参与吡啶环共轭有关。对于B3LYP方法,也具有同样的趋势,其中B3LYP/6-31G(d)方法的结果更为合理,在此方法中,吡虫啉的键长平均偏差仅为0.003 4 Å,化合物2仅为0.004 6 Å,二者的最大偏差均小于0.020 0 Å(吡虫啉的C1–Cl为0.018 2 Å,化合物2的C4–C5为0.019 1 Å);因此,可以认为使用的基组越大,其计算结果越精确,越接近试验值,所得的分子优势构型更为准确可靠。
为了更直观地显示键长理论计算值与试验值的相关性,以试验值为x轴,计算值为y轴,对它们
进行线性回归。统计分析结果表明,相关系数的平方(R2)能很好地反映各种优势构型与晶体结构的符合程度。表3结果表明,对于吡虫啉,在B3LYP/6-31G(d)水平上,R2为0.992 5,可见它们之间的相关性良好。AMl的相关性系数仅为0.725 4。而对于RHF和B3LYP方法,大基组计算结果比小基组计算结果精度高,与试验值相关性更好;因此,这些方法在优化吡虫啉时几何键长表现出的优劣顺序是:B3LYP/6-31G(d)、RHF/6-31G(d)、B3LYP/6-31G、RHF/6-31G、AM1。分析化合物2键长试验值与理论值的相关性时发现,基组和方法对分子构型的影响趋势与吡虫啉基本一致。AM1方法最为粗劣,B3LYP/6-31G(d)得出的优势构型与晶体结构符合程度最高。

表3 不同方法得到的键长理论值和试验值的相关系数Table 3 Correlation coefficients between the theoretical and the experimental bond lengths in different methods
2.3 不同量化方法对键角的影响
不同方法和基组计算的键角值和试验值列于表4、表5。结果表明,AMl方法计算的大部分键角与试验值差别都很大,只有个别键角与试验值相近。这说明AMl方法由于理论不严谨存在可信度差的缺点。RHF和B3LYP要优于AM1方法,但个别键角偏差较大,在2°~3°左右。这种偏差很大程度上可能是因为实验测量的晶体结构包含了较强的分子间相互作用,而量子化学计算出的是气态下的孤立分子结构。由于分子中键角弯曲振动模式的力常数一般总是比伸缩模式小得多,因此键角优化的误差对总能量的影响是一个次要因素。所以这种情况的偏差应该不会对一般分子的计算带来较大的误差。从相关系数(表6)来看,对于同一基组,同一方法(RHF或B3LYP),从小基组到大基组,相关性增大。其中B3LYP/6-31G(d)得出的相关系数最大,吡虫啉的R2为0.957 3,化合物2的R2为0.945 7,所得结论最为可靠。键角的统计分析结果与键长的情形相仿;因此,对于这3种理论方法而言,无论是吡虫啉或是化合物2,AMl法最粗略,B3LYP/6-31G(d)方法可信度最高,是新烟碱类杀虫剂分子构型优化最好方法。

表4 不同方法优化所得化合物1主要键角的理论值和试验值Table 4 Bond angles of compound 1 in different methods (°)
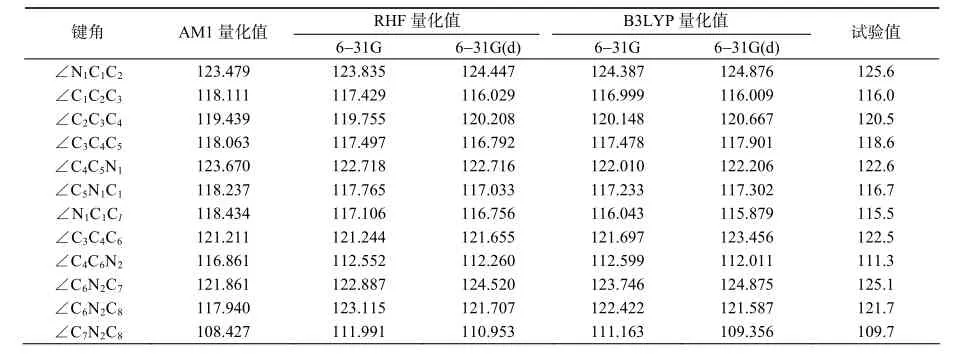
表5 不同方法优化所得化合物2主要键角的理论值和试验值Table 5 Bond angles of compound 2 in different methods (°)

续表

表6 不同方法得到键角理论值和试验值的相关系数Table 6 Correlation coefficients between the theoretical and the experimental bond Angles in differentm ethods
综合考虑键长和键角,AMl计算量虽较小,但由于它做了不同程度近似因而结果并不可靠;RHF方法没有考虑电子的相关效应,引起不可忽视的误差;B3LYP方法有效弥补了这一点,从而优化结果最为精确,有着其他方法不可比拟的优越性。特别是考虑了d轨道极化的B3LYP/6-31G(d)方法计算结果和试验值十分吻合。
2.4 不同量化方法对二面角的影响
通过对2种目标化合物晶体结构的比较分析发现,二者的二面角参数存在相当大的差异,这种差异主要体现在几个反映2种目标化合物硝基亚胺平面与相连杂环平面共面性的二面角上,化合物1的二面角N2—C7—N4—N5、N3—C7—N4—N5、C7—N4—N5—O1、C7—N4—N5—O2分别为176.3′、-3.0′、0.0′、179.4′,而化合物2与之对应的二面角为67.2′、-121.7′、-171.3′、8.5′。由此可看出,吡虫啉的亚硝基亚胺平面与相连杂环平面的共面性大大强于化合物2,而吡虫啉的活性(log(1/Ki)=6.00)大大高于化合物2(log(1/Ki)=3.86)[12]。由此可得出,新烟碱化合物分子中的强负电性基团所处的平面和与之相连的杂环平面之间的共面性对杀虫活性有非常要的影响。这些强负电性基团包括硝基亚胺基(—C==NNO2)、硝基亚甲基(—C==CHNO2)、氰基脒(—C—NCN)等。这与Tomizawa等[13]的观点是一致的。鉴于此,在选择计算方法时,进一步考察不同方法对目标化合物二面角参数的影响显得十分重要。
不同方法和基组计算的二面角值和试验值列于表7、表8。不难看出,各方法和基组对二面角参数的影响与键长、键角是一致的。鉴于上述结果,只着重分析B3LYP/6-31G(d)方法所得到的各二面角计算值与试验值的符合情况。由表7、表8可知,在B3LYP/6-31G(d)方法下,吡虫啉的二面角计算值的平均偏差仅为0.678′,4个主要二面角N2—C7—N4—N5、N3—C7—N4—N5、C7—N4—N5—O1、C7—N4—N5—O2的偏差分别为0.452′、0.084′、0.507′、0.467′,均小于平均偏差。化合物2的二面角计算值的平均偏差略大,为1.110′,4个主要二面角N2—C7—N4—N5、N3—C7—N4—N5、C7—N4—N5—O1、C7—N4—N5—O2的偏差分别为0.662′、0.216′、0.068′、1.433′。整体上,用B3LYP/6-31G(d)方法得到的二面角结果与试验值十分吻合。

表7 不同方法优化所得化合物1主要二面角的理论值和试验值Table 7 Theoretical and the experimental parameter in different methods of compound 1 (°)

表8 不同方法优化所得化合物2主要二面角的理论值和试验值Table 8 Theoretical and the experimental parameter in different methods of compound 2 (°)
2.5 各种方法优化后单点能的比较分析
从能量的角度来考察各种方法的优化效果,各种方法优化后的单点能列于表9。从表9可见,由于B3LYP理论考虑了电子相关能效应,因此选择各种基组的计算能量值都低于RHF,且随着基组的增大,能量降低。从能量角度考虑在B3LYP理论水平上,选择6-31G(d)基组进行构型优化,也是一个获得合理计算结果的方法。
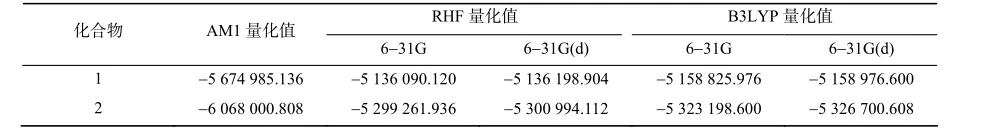
表9 不同水平和基组优化的目标化合物单点能Table 9 Single energy of compounds in different method J/mol
2.6 红外光谱分析
从图1中可看出,IMI分子有7个强特征峰,不同方法计算所得相应的7个特征峰波数列于表10。用B3LYP/6-31G(d)方法得到的IMI的7个强特征峰波数与试验值最接近,差值最小。为更直观反映不同方法对新烟碱类化合物红外光谱的影响,笔者计算了不同方法下7个强特征峰波数的相对误差(相对误差=((计算值-实验值)/实验值×100%)及平均值,结果列于表11。由表11可知,B3LYP/6-31G(d)方法的相对误差最小。在B3LYP理论水平上,选择6-31G(d)基组对新烟碱类化合物进行构型优化,所得的理论构型与真实构型最为吻合,本研究3种方法相比较,B3LYP/6-31G(d)是新烟碱类化合物量子

图1 ⅠMⅠ标准品的红外光谱Fig.1 ⅠR spectrum of ⅠMⅠ
化学研究的最佳方法选择。
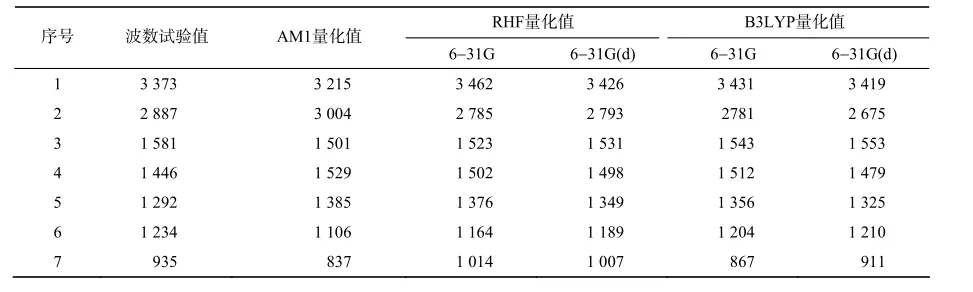
表10 ⅠMⅠ 7个强特征峰波数的试验值与理论计算值Table 10 Experienced and calculated wavenumbers of ⅠMⅠ cm-1
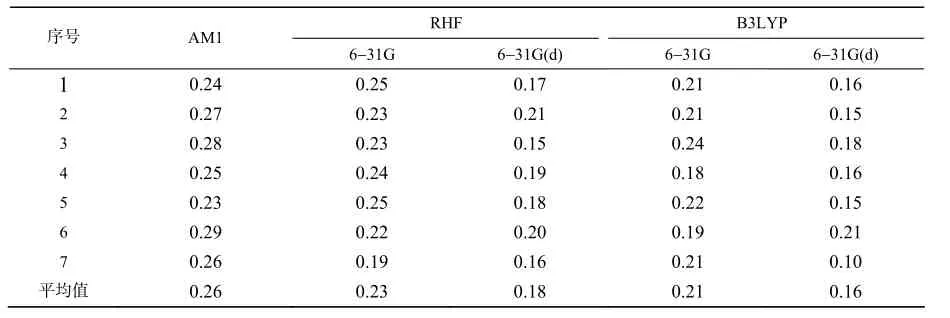
表11 ⅠMⅠ 7个强特征峰波数理论计算值的相对误差Table 11 Statistical error experienced and calculated wavenumbers of ⅠMⅠ %
2.7 3种方法所需CPU时间的比较
表12列出了不同方法优化和单点计算目标化合物各自所需的CPU时间。由于计算体系比较大,对称性低,加之分子数量较多,需要考虑计算所耗费的时间成本。由表12可知,当计算机内存有限,同时又要保证计算精度的前提下,B3LYP/6-31G(d)无疑是一种省时可行的方法。
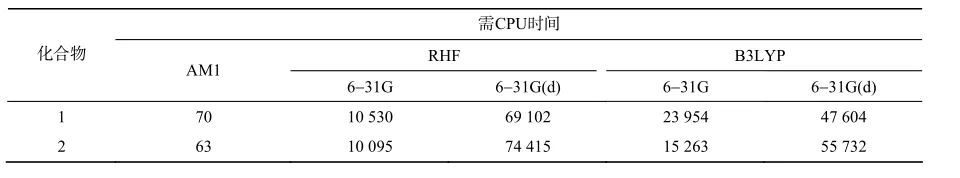
表12 不同方法所需的CPU时间Table 12 The CPU time of different methods s
[1] 唐振华.新烟碱型杀虫剂的结构与活性及其药效基团[J].现代农药,2002(1):1-6.
[2] 郭少雄,邢媛媛,王有名.新烟碱类似物构效关系的研究进展[J].精细化工中间体,2007,37(2):6-8,36.
[3] 唐敖庆,李前树.分子反应动力学[M].长春:吉林大学出版社,1989.
[4] 陈志行.有机分子轨道理论[M].济南:山东科学技术出版社,1991.
[5] Kagabu S,Matsuno H.Chloronicotinyl insecticides crystal and molecular structures of imidacloprid and analogous compounds[J].J Agric Food Chem,1997,45:276-281.
[6] Pastor M,Cruciani G,McLay I,et al.GRid-independent descripors(GRIND):Novel class of alignment-independent three-dimensional molecular descriptors[J].J Med Chem,2000,43(17):3233-3243.
[7] 徐光宪,黎乐民,王德民.量子化学——基本原理和从头计算法[M].北京:科学出版社,2009.
[8] 肖慎修,王崇愚,陈天朗.密度泛函理论的离散变分方法在化学和材料物理学中的应用[M].北京:科学出版社,1998.
[9] Slater J C.A simplification of the hartree fock method [J].Phys Rev,1951,81:385-390.
[10] 王莹.4种喹诺酮类药物分子荧光光谱的量子化学研究[J].四川理工学院学报:自然科学版,2010,23(5):570-571,579.
[11] 张姝,陈国力,刘姗,等.三种农药荧光光谱的量子化学研究[J].光谱学与光谱分析,2009,29(1):169-171.
[12] Christof F S, Daniel L C, Thomas C S,et al. Insecticidal activity and mode of action of novel nicotinoids synthesized by new acylpyridinium salt chemistry and directed lithiation pesticide[J]. Biochemistry and Physiology, 2007,87: 211-219.
[13] Tomizawa M,Lee D L,Casida J E.Neonicotiiloid insecticides:Molecular features conferring selectivity for insect versus mammalian nicotinic receptor[J].Agric Food Chem,2000,48:6016-6024.
