HPLC法测定头孢泊肟酯分散片中有关物质的含量
2011-02-13南京市溧水县人民医院溧水县211200
俞 平(南京市溧水县人民医院,溧水县 211200)
头孢泊肟酯(Cefpodoxime Proxetil)为第3代广谱半合成口服头孢菌素,为头孢泊肟的前体药物。头孢泊肟酯本身无抗菌活性,在肠壁被非特异性的酯酶水解成头孢泊肟而发挥抗菌活性。头孢泊肟有广谱而强大的抗菌作用,组织分布广泛,具有治疗量小、每日给药频数少的优点[1]。分散片是能迅速崩解的一种速释剂型,主要用于溶解性差的药物。头孢泊肟酯极易溶于甲醇或乙腈,易溶于乙醇,几乎不溶于水[2]。笔者采用高效液相色谱(HPLC)法对自制的头孢泊肟酯分散片进行了有关物质检测,并进行方法学研究。
1 仪器与试药
LC-10A高效液相色谱仪、SPD-10A紫外检测器(日本岛津公司);N2010色谱工作站(浙江大学智能信息研究所);UV-2450紫外-可见分光光度计(日本岛津公司)。
头孢泊肟酯对照品(中国药品生物制品检定所,批号:130517-200401);头孢泊肟酯分散片(自制,规格:每片含头孢泊肟0.1 g,批号:101010、101011、101012);甲醇为色谱纯,水为纯化水。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:ODS(C18)柱(250 mm×4.6 mm,5 μm);流动相:水-甲醇(55∶45);检测波长:240 nm;流速:2.0 mL·min-1;进样量:20 μL;柱温25℃。头孢泊肟酯同分异构体A、B间分离度应≥4.0,头孢泊肟酯异构体B理论板数应≥5 000。在此条件下头孢泊肟酯与有关杂质实现基线分离。色谱见图1。
2.2 溶液的制备
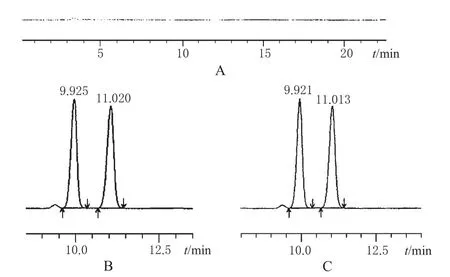
图1 高效液相色谱图A.空白溶液;B.对照品溶液;C.样品溶液Fig 1 HPLC chromatogramsA.blank solution;B.control solution;C.sample solution
2.2.1 供试品溶液的制备 取本品20片,精密称定,研细,精密称取适量(约相当于头孢泊肟酯100 mg),置于100 mL容量瓶中,加流动相溶解并稀释至刻度,制成每1 mL中含头孢泊肟酯1 mg的溶液,摇匀,过滤,作为供试品溶液。
2.2.2 对照品溶液的制备 精密称定头孢泊肟酯对照品约100 mg,置于100 mL容量瓶中,加流动相溶解并稀释至刻度,制成每1 mL中含头孢泊肟酯1 mg的溶液,摇匀,过滤,作为对照品溶液。
2.2.3 对照溶液的制备 精密量取供试品溶液1 mL,置于100 mL容量瓶中,加流动相稀释至刻度,作为对照溶液。
2.3 辅料干扰试验
根据处方比例及生产工艺制备空白片,取空白片20片,精密称定,研细,精密称取0.5 g,置于100 mL容量瓶中,加流动相溶解并稀释至刻度,得空白对照溶液。取样品20片,精密称定,研细,精密称取0.5 g,按照“2.2.1”项下方法制备供试品溶液。按照“2.1”项下方法分别测定空白对照、供试品溶液。结果,混合辅料溶液在该色谱条件下无吸收,故辅料不干扰本品有关物质的检测。
2.4 空白溶剂试验
取流动相20 μL注入液相色谱仪,按照“2.1”项下方法测定。结果,溶剂在该色谱条件下无吸收,故空白溶剂不干扰本品的有关物质检测。
2.5 破坏试验
2.5.1 强碱破坏试验 取本品10片,精密称定,研细,精密称取适量(约相当于头孢泊肟酯50 mg),置于50 mL容量瓶中,加入0.5 N氢氧化钠溶液10 mL,于室温条件下破坏2 h,再用0.5 N盐酸调节溶液pH至中性,再加流动相稀释至刻度,滤过,取续滤液按照“2.1”项下方法测定。结果,主要降解产物的保留时间为10 min和15.2~16.4 min,各降解产物与头孢泊肟酯A(11.5 min)、头孢泊肟酯B(14.4 min)及头孢泊肟酯A和B中间的Δ3-异构体(12.4 min)有较好的分离效果。
2.5.2 强酸破坏试验 取本品10片,精密称定,研细,精密称取适量(约相当于头孢泊肟酯50 mg),置于50 mL容量瓶中,加入0.5 N盐酸溶液10 mL,于室温条件下破坏2 h,再用0.5 N氢氧化钠调节溶液pH至中性,再加流动相稀释至刻度,滤过,取续滤液按照“2.1”项下方法测定。结果,主要降解产物的保留时间为0~6 min及10 min,头孢泊肟酯A、头孢泊肟酯B及头孢泊肟酯A和B中间的Δ3-异构体基本完全破坏。
2.5.3 强光照射破坏试验 取本品10片,裸片放置在光照强度为(4 500±500)lx的条件下破坏48 h,精密称定,研细,精密称取适量(约相当于头孢泊肟酯50 mg),置于50 mL容量瓶中,加流动相溶解并稀释至刻度,滤过,取续滤液按照“2.1”项下方法测定。结果,主要降解产物的保留时间为10 min和15.2~16.4 min,各降解产物与头孢泊肟酯A(11.5 min)、头孢泊肟酯B(14.4 min)及头孢泊肟酯A和B中间的Δ3-异构体(12.4 min)有较好的分离效果。
2.5.4 高温破坏试验 取本品10片,裸片放置在105℃烘箱中破坏48 h,精密称定,研细,精密称取适量(约相当于头孢泊肟酯50 mg),置于50 mL容量瓶中,加流动相溶解并稀释至刻度,滤过,取续滤液按照“2.1”项下方法测定。结果,主要降解产物的保留时间为10 min和15.2~16.4 min,各降解产物与头孢泊肟酯A(11.5 min)、头孢泊肟酯B(14.4 min)及头孢泊肟酯A和B中间的Δ3-异构体(12.4 min)有较好的分离效果。
2.5.5 过氧化氢破坏试验 取本品10片,精密称定,研细,精密称取适量(约相当于头孢泊肟酯50 mg),置于50 mL容量瓶中,加入30%过氧化氢10 mL,于室温条件下破坏2 h,加流动相溶解并稀释至刻度,滤过,取续滤液按照“2.1”项下方法测定。结果,主要降解产物的保留时间为0~6、10和15.2~16.4 min,各降解产物与头孢泊肟酯A(11.5 min)、头孢泊肟酯B(14.4 min)及头孢泊肟酯A和B中间的Δ3-异构体(12.4 min)有较好的分离效果。
2.6 最小检出量
取头孢泊肟酯对照品溶液,加流动相振摇使溶解并制成一定浓度的溶液,并逐级稀释,取20 μL注入液相色谱仪,根据信噪比(S/N=3),头孢泊肟酯A的最小检测限为1.6 ng,头孢泊肟酯B的最小检测限为1.2 ng。
2.7 重复性试验
精密称取批号为101010的头孢泊肟酯分散片6份,研细,精密称定每份0.50 g(约含头孢泊肟酯100 mg),置于100 mL容量瓶中,按照“2.2”项下方法制备供试品溶液和对照品溶液,按照“2.1”项下方法进行有关物质测定。结果,有关物质的平均含量为3.03%,RSD=0.3%;Δ3-异构体为3.12%,RSD=0.4%,表明该方法重复性良好。
2.8 精密度试验
取头孢泊肟酯对照品适量,按“2.2.1”项下方法制备对照品溶液,按“2.1”项的方法连续6次进样测定,记录色谱图。结果,RSD=0.51%,表明该方法精密度较高。
2.9 头孢泊肟酯分散片有关物质测定
通过方法学研究试验,表明上述有关物质的测定方法可行。取自制的3批头孢泊肟酯分散片,分别制备供试品溶液和自身对照溶液,进行3批样品的有关物质的检测。结果,有关物质分别为3.07%、2.92%、3.1%;Δ3-异构体分别为3.24%、3.08%、3.04%,表明试验结果均符合规定。
3 讨论
3.1 测定波长的选择
取头孢泊肟酯对照品适量,精密称取10 mg,置于100 mL容量瓶中,加甲醇溶解并稀释至刻度,摇匀,取续滤液1 mL,置于10 mL容量瓶中,加甲醇稀释至刻度,摇匀,在200~400 nm波长处扫描。结果,头孢泊肟酯溶液在240 nm处有最大吸收,故选择240 nm作为检测波长。
3.2 流动相的选择
参照2010年版《中国药典》(二部)中头孢泊肟酯质量标准中有关物质项下的色谱方法,分别对水-甲醇[2]及0.01 mol·L-1的醋酸溶液-乙腈[3]流动相系统进行筛选,在不同流动相条件及比例下,分别取供试品溶液进样,记录色谱图。试验结果表明,在0.01 mol·L-1的醋酸溶液-乙腈系统条件下,主峰峰形较差;水-甲醇系统,在水与甲醇比例为55∶45时,主峰峰形好,保留时间合适,头孢泊肟酯A和B间分离度>4.0,头孢泊肟酯异构体B>5 000。故确定水-甲醇(55:45)作为流动相。
3.3 破坏性试验
破坏性试验是在人为设定的特殊条件下,使药物降解,来验证方法的可行性,分析药物可能的降解途径和降解产物。破坏性试验的条件通常有:酸——0.1~1 mol·L-1盐酸或硫酸溶液;碱——0.1~1 mol·L-1的氢氧化钠溶液;氧化——适当浓度的过氧化氢溶液;高温——通常温度高于加速试验温度;强光——可采用4 500 lx强光照射。具体条件需结合药物的特点,使药物有一定量的降解。本试验采用了适宜的破坏方法,在这些条件下,头孢泊肟酯均有一定的降解,且降解产物与主成分峰均有较好的分离效果。
综上所述,通过多次试验确定头孢泊肟酯分散片有关物质检查的方法和条件,在此条件下辅料及溶剂对头孢泊肟酯的有关物质测定无干扰,有关物质和降解产物与主成分峰分离效果良好;并对该方法进行方法学考察,表明该法简便、快速、准确,可用于头孢泊肟酯质量控制。
[1] 邵长周,瞿介明,何礼贤.第3代头孢菌素——头孢泊肟酯[J].中国新药与临床杂志,2004,23(11):746.
[2] 国家药典委员会编.中华人民共和国药典(二部)[S].2010年版.北京:中国医药科技出版社,2010:198-200.
[3] 日本药局方编辑委员会编.日本药局方[S].15 XV.2005:461-462.
