心肌炎小鼠心肌纤维化与心脏上皮/内皮间充质转化的关系*
2011-01-30张召才虞意华颜默磊吕晓春
张召才, 严 静, 虞意华, 颜默磊, 吕晓春
(浙江医院ICU,浙江 杭州310013)
心肌纤维化是病毒性心肌炎常见病理变化,是患者出现心律失常、心功能减退甚至心脏性猝死等并发症的重要原因[1]。既往认为心脏原位(heartresident)的成纤维细胞(fibroblast,Fb)受炎症等刺激后增生、活化产生胶原和非胶原性细胞外基质(extracellular matrix,ECM)是心肌纤维化形成的核心事件。近年来研究表明:心脏成纤维细胞(cardiac fibroblast,cFb)有多个来源,除心脏原位cFb外,骨髓和心脏组织中的上皮/内皮细胞均是 cFb的来源[2-4],后者通过上皮/内皮间充质转化(epithelial/endothelial to mesenchymal transition,EMT/EndMT)可以转变成为cFb[2,5]。心外来源的cFb与心肌纤维化的关系日益受到关注,在自身免疫性心肌炎模型中发现骨髓来源的 cFb约占所有 cFb的60%[3,4];目前尚不清楚EMT/EndMT是否与病毒性心肌炎心肌纤维化有关,本文对此进行研究和探讨。
材料和方法
1 试剂和仪器
1.1 试剂 天狼星红(Sirius red,F3BA,Fluka Chemie);转化生长因子(transforming growth factor β1,TGF- β1)、Wnt1、Twist1、血管内皮细胞钙黏素(VE-cadherin)、上皮细胞钙黏素(E-cadherin)、成纤维细胞特异蛋白(fibroblast specific protein-1,FSP-1)、α-平滑肌肌动蛋白(α -smooth muscle actin,α-SMA)和内参照α-微管蛋白(α-tubulin)Ⅰ抗均购自 Santa Cruz。Ⅱ抗购自 Jackson。胶原前肽ELISA试剂盒购自上海蓝基生物科技公司。Trizol试剂盒购自Invitrogen。Western blotting ECL试剂盒购自Pirece。PVDF免疫印迹转移膜购自Millipore。逆转录试剂盒购自Promega。聚丙烯酰胺凝胶购自上海碧云天生物公司。
1.2 主要仪器和软件 ABI Prism 7700序列测定仪(ABI);HRD-045-NIK尼康照相系统(尼康);凝胶扫描分析系统(Total lab)。
2 方法
2.1 建立动物模型 雄性、6周龄、纯种近系BALB/c小鼠36只(购自上海斯莱克实验动物公司,平均体重20-25 g,随机分为2组:对照组(control,n=16)和病毒性心肌炎组(viral myocarditis,VMC,n=20)。对照组每周腹腔注射1次无菌病毒培养液(EMEM)0.1 mL;心肌炎组每周腹腔接种1次0.1 mL 109倍50%组织培养感染剂量(50%tissue culture infection dose,TCID50)柯萨奇病毒B3(Coxsackievirus B3,CVB3,Nancy株),复制急、慢性病毒性心肌炎模型。7 d后从2组中随机选取活鼠各8只处死取样;余小鼠继续腹腔注射和自然喂养直至第30 d,统计最后存活小鼠数。
2.2 标本处理 在上述各组小鼠达到观察时点时,随机取心肌炎组与对照组小鼠各8只,行心脏超声检查心功能,然后处死小鼠。所有存活小鼠均取血并提取血清置入-20℃冰箱冻存待测;心脏组织切成2块,分别用于组织病理学检测(10%中性甲醛PBS固定)、基因检测(-70℃冻存)和蛋白检测(-40℃冻存)。
2.3 组织病理学检测 甲醛固定的标本经脱水、清洗后以石蜡包埋,其后制成0.4μm厚切片,常规HE染色并行Picrosirius red胶原特异染色后镜检。拍片后,取8个不重复视野以自动化照相系统定量分析病变程度和计算胶原容积分数(collagen volume fraction,CVF)。
2.4 血清学检测 应用ELISA检测如下胶原前肽:III型前胶原氨基端前肽(aminoterminal propeptide of typeⅢ procollagen,PⅢNP)、I型前胶原氨基端前肽(aminoterminalpropeptide oftype Iprocollagen,PINP)和 I型前胶原羧基端前肽(carboxyterminal propeptide of type I procollagen,PICP),以评价心脏组织中I型和Ⅲ型胶原代谢情况。操作严格参照说明书进行,主要步骤包括:标本激活、建立标准曲线、加样、加酶标抗体、加底物工作液、终止液和显色等,最后在492 nm处测吸光度值(A值),根据各样品A值在标准曲线上查出所测指标的含量。
2.5 实时RT-PCR检测 从每组随机选择6份心肌标本,以Trizol试剂提取总RNA,采用紫外分光光度法测定RNA样品在波长260 nm和280 nm的紫外吸收值,计算 A260/A280确认其纯度和浓度。取2 μg总RNA应用逆转录试剂盒两步法合成cDNA第1链;将所得cDNA稀释30倍后取3 μL分别加入预先合成的前向和逆向引物各1 μL和5 μL subgreen mix组成10 μL实时RT-PCR反应体系,RT-PCR程序为:50℃ 2 min,95 ℃ 10 min,95 ℃ 15 s,60 ℃1 min,95℃ 15 s,共40个循环。反应结束后确认real-time PCR的扩增曲线和融解曲线,计算Ct值(threshold cycle),采用比较Ct值方法以β-actin为内参照相对定量各检测基因的表达水平。每一实验至少重复3次。所有引物均委托上海生工生物公司设计和合成,见表1。
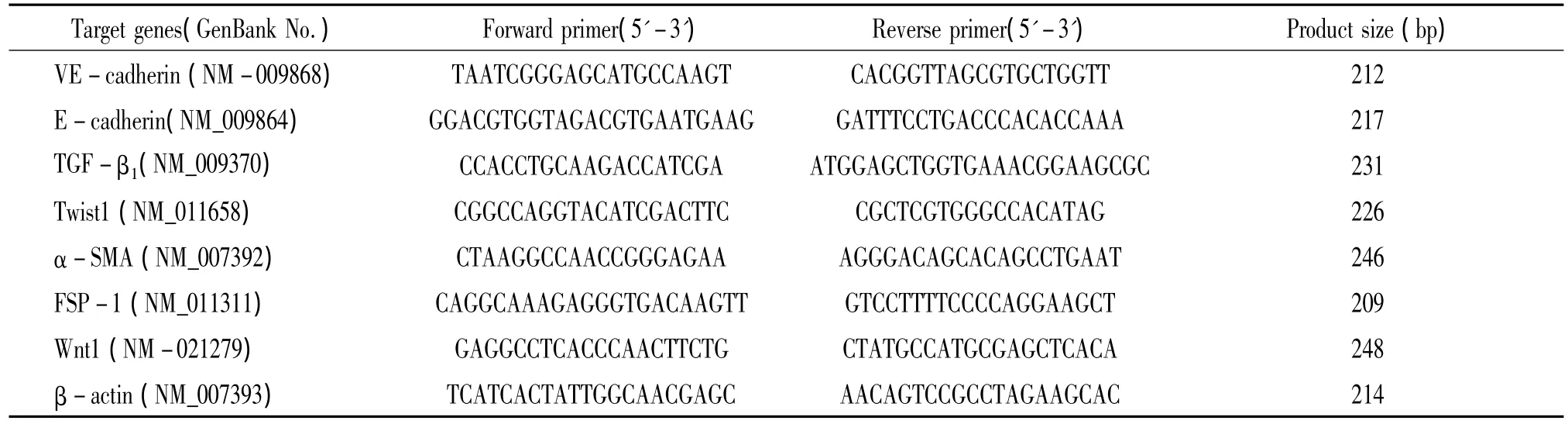
表1 检测目标基因所有引物序列Table 1.Primer sequences for detection of target genes
2.6 Western blotting检测 以Trizol试剂盒抽提心肌样品总蛋白,各取10 μL蛋白样品进行聚丙烯酰胺凝胶电泳,印迹转染PVDF膜。用含5%脱脂奶粉的0.05%Tween-20/PBS封闭液在室温下孵育1 h以去除非特异结合部份,封闭液中分别加入2 μL抗 TGF-β1、Wnt1、Twist1、VE-cadherin、E-cadherin、FSP-1、α-SMA和内参照α-tubulin抗体,4℃过夜。加入3 μL第Ⅱ抗体后,用Western blotting ECL试剂盒检测免疫复合物,照相显影后经Total Lab 1.11凝胶扫描分析系统进行灰度扫描并作半定量分析。
3 统计学处理
结 果
1 小鼠存活率与组织病理学检查结果
除外第7 d处理的小鼠(每组8只,共16只),第30 d,剩余小鼠中心肌炎组存活9只(9/12),对照组8只均存活(8/8)。HE染色:第7 d,局部心脏可见炎症细胞浸润,心肌细胞坏死,心肌纤维断裂;第30 d,炎症细胞数减少,纤维组织替代部分坏死心肌,心肌细胞体积增大,排列紊乱,核大深染、异形,心肌纤维基本完整。此病理改变提示急、慢性病毒性心肌炎模型成功建立并均出现心肌纤维化,与我们此前报道的结果一致[6]。
2 血清胶原前肽检测结果
心肌炎组小鼠血清胶原前肽产生明显增高,与CVF变化相一致,提示急、慢性心肌炎小鼠均出现心肌纤维化,见表2。
3 急性心肌炎心脏中发生EMT/EndMT
在基因和蛋白表达水平均证实急性心肌炎(第7 d)小鼠心脏中发生EMT/EndMT,特征为上皮/内皮细胞表型标志(E-cadherin和VE-cadherin)丢失,表达明显下调;间充质蛋白(FSP-1和α-SMA)产生增多,表达显著上调,伴诱导性细胞因子 TGF-β1、转录因子Wnt1、Twist1等表达增加,同时胶原合成增强(CVF和胶原前肽升高),与心肌纤维化变化一致,见表2、3和图1。
4 慢性心肌炎心肌中未再发现EMT和EndMT
在基因和蛋白水平均证实慢性感染(第30 d)小鼠心脏中未再发现EMT/EndMT,虽然 FSP-1和α-SMA 的表达仍然显著上调,TGF-β1、Wnt1、Twist1等因子表达增加,心肌纤维化仍然明显,但E-cadherin和VE-cadherin表达水平未见下调,表2、3和图2。
表2 小鼠血清胶原前肽和心脏胶原容积分数的比较Table 2.Comparison of serum procollagen propeptides and cardiac collagen volume fraction(CVF)in mice(±s)
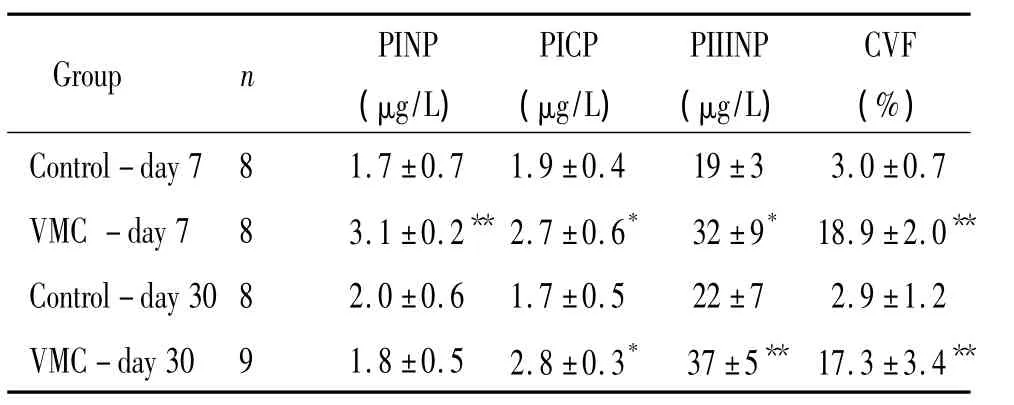
表2 小鼠血清胶原前肽和心脏胶原容积分数的比较Table 2.Comparison of serum procollagen propeptides and cardiac collagen volume fraction(CVF)in mice(±s)
*P <0.05 ,**P <0.01 vs control.
Control-day 7 8 1.7 ±0.7 1.9 ±0.4 19 ±3 3.0 ±0.7 VMC-day 7 8 3.1 ±0.2** 2.7 ±0.6* 32 ±9* 18.9 ±2.0**Control-day 30 8 2.0 ±0.6 1.7 ±0.5 22 ±7 2.9 ±1.2 VMC-day 30 9 1.8 ±0.5 2.8 ±0.3* 37 ±5** 17.3 ±3.4**
讨 论
EMT是胚胎期器官形成发育过程中的重要事件,EndMT是其特殊表现形式,近年来发现EMT/EndMT参与了病理性器官纤维化,包括肾、肺、肝、肠和心肌纤维化等[2,5,7,8]。肾损伤后纤维化时肾脏 Fb有 14%-15%来自骨髓、36%经由 EMT 而来[7,8];在压力超负荷和慢性移植排斥大鼠模型中发现:27%-35%心脏Fb经由EndMT而来[2]。本研究发现,仅在急性病毒感染时小鼠心脏同时出现EMT和EndMT,伴心肌纤维化形成;慢性病毒感染时,心肌纤维化依然明显,但未见EMT和EndMT伴随。
急性病毒感染时小鼠心脏出现EMT和EndMT的原因考虑与心脏上皮/内皮细胞屏障受损、细胞连接破坏有关。上皮/内皮受损引起TGF-β1、Wnt1、Twist1等的表达上调,可以促使上皮/内皮细胞向间充质细胞的转变、加速胶原的形成,有利于限制炎症反应、减轻组织器官损伤[8,9],因此急性期心肌纤维化常常被认为是有益的修复性纤维化,此时胶原的合成和降解同时增强,有利于使心脏炎症局限化[6]。有人认为急性期心肌纤维化不宜干预,但在临床实践中发现:治疗急性心肌炎时,控制炎症反应、保护心肌等措施往往也会减轻心肌纤维化,与心肌纤维化有关的心力衰竭、心律失常、心脏性猝死等并发症的发生率均会下降[10],从这一点看来似乎有又干预的必要。本研究结果给干预急性心肌炎带来了新的思路,在治疗急性心肌炎时,除采取抗感染、抗病毒、免疫抑制、抗心衰、抗心律失常等[10]之外,如果增加上皮/内皮细胞保护和干细胞治疗等措施,有可能进一步改善预后和减轻纤维化,减少近期和远期并发症的发生[11,12],但其临床有效性和安全性需要进一步验证。
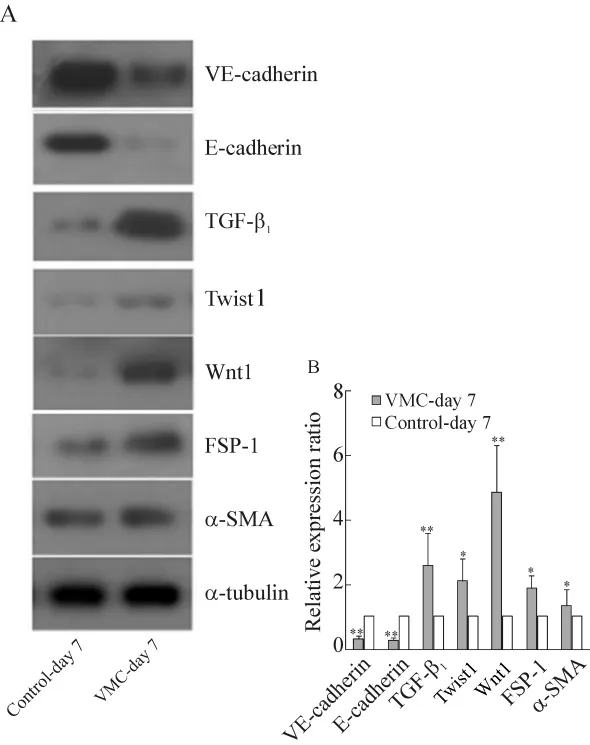
Figure 1.Cardiac EMT/EndMT proteomes expression in mice with acute viral myocarditis.A:expression of EMT/EndMT proteomes detected by Western blotting;B:relative expression ratio of EMT/EndMT proteomes between the 2 groups.±s.n=6.*P<0.05,**P<0.01 vs control.图1 急性病毒性心肌炎小鼠心脏EMT/EndMT蛋白质组的表达
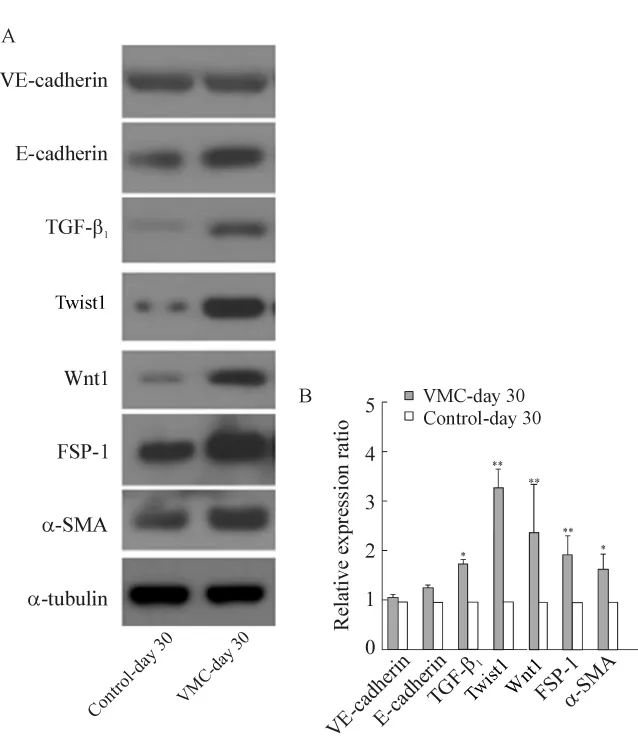
Figure 2.Cardiac EMT/EndMT proteomes expression in mice with chronic viral myocarditis.A:expression of EMT/EndMT proteomes detected by Western blotting;B:relative expression ratio of EMT/EndMT proteomes between the 2 groups.±s.n=6.*P<0.05,**P <0.01 vs control.图2 慢性病毒性心肌炎小鼠心脏中EMT/EndMT蛋白质组的表达
表3 急、慢性期2组小鼠EMT/EndMT相关基因表达水平的比较Table 3.Comparison of relative mRNA expression levels of EMT/EndMT-related genes between the 2 groups of mice in acute and chronic stage(±s.n=6)
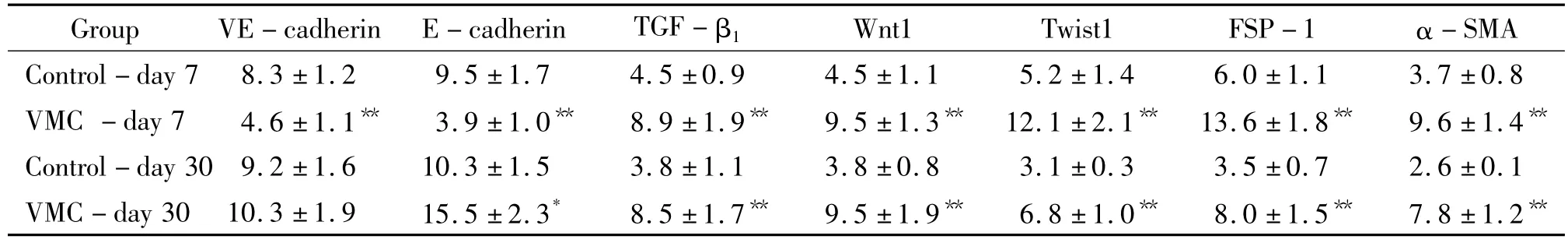
表3 急、慢性期2组小鼠EMT/EndMT相关基因表达水平的比较Table 3.Comparison of relative mRNA expression levels of EMT/EndMT-related genes between the 2 groups of mice in acute and chronic stage(±s.n=6)
*P <0.05,**P <0.01 vs control.
Group VE-cadherin E-cadherin TGF-β1 Wnt1 Twist1 FSP-1 α-SMA Control-day 7 8.3 ±1.2 9.5 ±1.7 4.5 ±0.9 4.5 ±1.1 5.2 ±1.4 6.0 ±1.1 3.7 ±0.8 VMC-day 7 4.6 ±1.1** 3.9 ±1.0** 8.9 ±1.9** 9.5 ±1.3** 12.1 ±2.1** 13.6 ±1.8** 9.6 ±1.4**Control-day 30 9.2 ±1.6 10.3 ±1.5 3.8 ±1.1 3.8 ±0.8 3.1 ±0.3 3.5 ±0.7 2.6 ±0.1 VMC-day 30 10.3 ±1.9 15.5 ±2.3* 8.5 ±1.7** 9.5 ±1.9** 6.8 ±1.0** 8.0 ±1.5** 7.8 ±1.2**
慢性期未出现EMT/EndMT可能是因为受损的内皮/上皮已经完成自我修复更新,此时TGF-β1、Wnt1、Twist1等的活跃表达可能通过影响心脏原位的或骨髓来源cFb的活动而促进心肌纤维化形成[8,10,11]。这一结果一方面说明慢性心肌炎心肌纤维化的发生机制与急性期有很大差异,另一方面提示对慢性期心肌纤维化的干预要寻找新的切入点。干预慢性心肌炎心肌纤维化时除了针对TGF-β1和Wnt1的刺激因素如病原体、神经体液因子、机械牵拉等[9]之外,还可以考虑采取措施直接抑制或拮抗TGF-β1和Wnt1,以及针对 TGF-β1/Smad通路和Wnt1/β-catenin信号通路中的重要信号分子进行干预。TGF-β1和Wnt1均是促纤维化的重要信号分子,TGF-β1可以调控间质细胞、炎症细胞和产胶原细胞的交互作用,在多种器官纤维化过程中起关键作用,抑制 TGF-β1可以明显改善器官纤维化[9,13];TGF- β1/Smad 和 Wnt1/β -catenin 两大信号通路系统在肺纤维化时已经发现有协同作用[14]。据此推测在我们的心肌炎模型中TGF-β1/Smad和Wnt1/β-catenin两大信号通路系统的关键因子TGF-β1和Wnt1同时上调也可能是一种协同效应,因此,采用针对这两大信号通路的干预很可能可以抑制或延缓心肌纤维化进程;但由于这些信号分子往往具有多种功能,针对它们的干预在实际操作时需要慎重考虑,以免引起不良后果。
[1]Massare J,Berry JM,Luo X,et al.Diminished cardiac fibrosis in heart failure is associated with altered ventricular arrhythmia phenotype[J].J Cardiovasc Electrophysiol,2010,21(9):1031-1037.
[2]Zeisberg EM,Kalluri R.Origins of cardiac fibroblasts[J].Circ Res,2010,107(12):1304-1312.
[3]Blyszczuk P,Kania G,Dieterle T,et al.Myeloid differentiation factor-88/interleukin-1 signaling controls cardiac fibrosis and heart failure progression in inflammatory dilated cardiomyopathy [J].Circ Res,2009,105(9):912-920.
[4]Kania G,Blyszczuk P,Stein S,et al.Heart-infiltrating prominin-1+/CD133+progenitor cells represent the cellular source of transforming growth factor β-mediated cardiac fibrosis in experimental autoimmune myocarditis[J].Circ Res,2009,105(5):462-470.
[5]Guarino M,Tosoni A,Nebuloni M.Direct contribution of epithelium to organ fibrosis:epithelial-mesenchymal transition [J].Hum Pathol,2009,40(10):1365-1376.
[6]张召才,杨英珍,陈瑞珍,等.病毒性心脏病小鼠心脏胶原代谢的动态变化[J].中国病理生理杂志,2006,22(8):1529-1534.
[7]Kalluri R,Weinberg RA.The basics of epithelial-mesenchymal transition[J].J Clin Invest,2009,119(6):1420-1428.
[8]Kisseleva T,Brenner DA.Mechanisms of fibrogenesis[J].Exp Biol Med,2008,233(2):109-122 .
[9]Arciniegas E,Frid MG,Douglas IS,et al.Perspectives on endothelial to mesenchymal transition:potential contribution to vascular remodeling in chronic pulmonary hypertension [J].Am J Physiol Lung Cell Mol Physiol,2007,293(1):L1-L8.
[10]Cooper LT Jr.Myocarditis[J].N Engl J Med,2009,360(15):1526-1538.
[11]Kania G,Blyszczuk P,Eriksson U.Mechanisms of cardiac fibrosis in inflammatory heart disease[J].Trends Cardiovasc Med,2009,19(8):247-252 .
[12]Ishikane S,Yamahara K,Sada M,et al.Allogeneic administration of fetal membrane-derived mesenchymal stem cells attenuates acute myocarditis in rats[J].J Mol Cell Cardiol,2010,49(5):753-761.
[13]Leask A.TGFβ,cardiac fibroblasts,and the fibrotic response[J].Cardiovasc Res,2007,74(2):207-212.
[14]Kim KK,Wei Y,Szekeres C,et al.Epithelial cell α3β1 integrin links β-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis[J].J Clin Invest,2009,119(1):213-224.
