HPLC法测定牛膝胶囊中β-脱皮甾酮的含量
2011-01-29陈玉敏水彩红
陈玉敏* 曹 红 水彩红
(总后卫生部药品仪器检验所,北京 100071)
牛膝胶囊由从牛膝提取物制成的胶囊剂,具有扶正补益,补肝肾,强筋骨[1-3]。用于预防和治疗因机体受到放射物质或化学物质损伤引起的白血球下降[4-7]。为了有效控制制剂质量,本文建立了HPLC法测定方中牛膝中β-脱皮甾酮的含量。
1 仪器与试药
仪器:岛津LC-20A高效液相色谱仪。试剂:甲醇为色谱纯级;水为双重蒸馏水。对照品:β-脱皮甾酮(批号:111638-200402中检所提供)。样品:牛膝胶囊(批号100101,100102,100103),自制。
2 实验方法与结果
2.1 色谱条件
色谱柱:SHIMADZU VP-ODS(5µm,150×4.6mm);流动相:甲醇-水(40∶60);检测波长:250nm(带宽4nm);流速:1.0mL/min;柱温:40℃。
2.2 对照品溶液及供试品溶液的制备
对照品溶液的制备:取β-脱皮甾酮对照品适量,精密称定,加甲醇制成每lmL含20μg的溶液,即得。
供试品溶液的制备:取装量差异项下的本品适量,研细,取0.5g,精密称定,置具塞三角瓶中,精密加甲醇25mL,称定重量,超声处理(功率300W,频率40kHz)30min,放冷,再称定重量,以甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.3 专属性试验(阴性对照试验)
阴性样品的制备:按处方比例组不含牛膝药材的方,按样品制备工艺制成阴性对照的制剂,再按供试品溶液制备方法处理,即得。
按样品测定的色谱条件进样,对照品、样品、阴性对照的液相色谱图1~3如下,阴性对照无干扰峰。
2.4 线性关系
取β-脱皮甾酮对照品制成不同浓度对照品溶液,分别精密吸取对照品溶液10mL,注入高效液相色谱仪,按样品测定色谱条件测定,以峰面积积分值为纵坐标,进样量(μg)为横坐标,绘制标准曲线,计算。
回归方程:Y=1589.9X-2848.9,相关系数r=1(n=5)。
结果表明β-脱皮甾酮在40~400mg范围内呈良好的线性关系。
2.5 精密度试验
精密吸取β-脱皮甾酮对照品溶液10μL,重复进样5次,测定,结果RSD分别为0.05%,表明精密度良好。
2.6 稳定性试验
取供试品溶液,在室温条件下,于0、2、4、6、8、10h,测定含量,β-脱皮甾酮测定结果的RSD为0.09%,表明样品溶液较稳定。
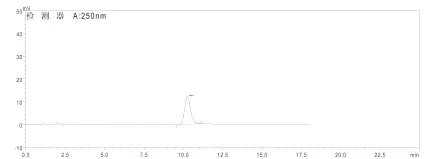
图1 β-脱皮甾酮对照品HPLC色谱图
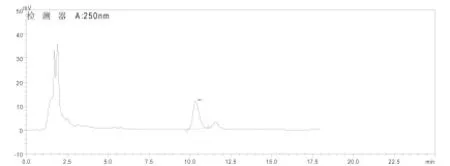
图2 样品HPLC色谱图
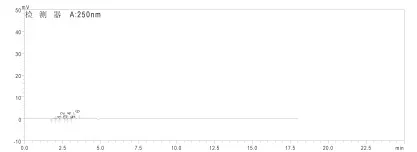
图3 阴性对照HPLC色谱图
2.7 重复性试验
取牛膝胶囊(100101),精密称取6份,平行操作,进行液相分析,样品中β-脱皮甾酮平均含量为:RSD为1.76%,具良好重复性。
2.8 回收率试验
采用加样回收,精密称取已知含量的牛膝胶囊(100101)0.25g,精确加入相当量的β-脱皮甾酮对照品,依法操作,测定牛膝胶囊中β-脱皮甾酮含量,计算回收率,测定结果见表1。

表1 样品中β-脱皮甾酮的加样回收率试验结果
2.9 样品测定
取牛膝胶囊3批,依法进行含量的测定,β-脱皮甾酮的含量结果见表2。
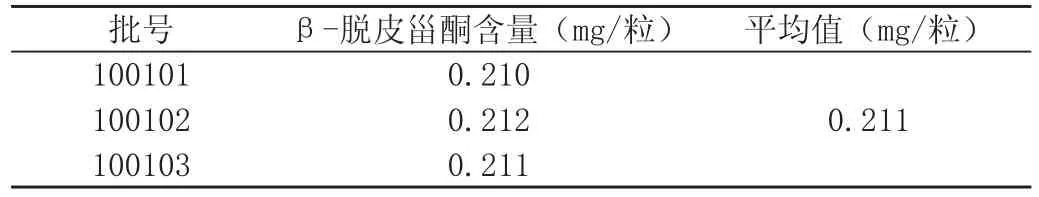
表2 牛膝胶囊中β-脱皮甾酮含量测定结果
3 讨 论
3.1 收集β-脱皮甾酮对照品及样品溶液光谱,在250nm处有最大吸收,因此选用250nm为检测波长。
3.2 试验选用甲醇、乙醇、无水乙醇作为提取溶媒进行比较,提取方法及含量测定方法相同,以甲醇为提取溶媒含量较高,选择甲醇作为测定牛膝中β-脱皮甾酮的溶剂。
3.3 提取方法分别采用超声提取15、30、45min,热回流提取30min,测定方法相同,以超声提取30min含量较高,选用甲醇作为提取溶媒、超声提取30min的提取方法作为测定牛膝中β-脱皮甾酮的方法。
[1]国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2010:67.
[2]李文莉,汪文涛,雷玉萍.RH-HPLC法测定脉络宁注射液中脱皮甾酮的含量[J].华西药学杂志,2001,16(4):382-383.
[3]张翠英,梁生旺,张广强.不同产地牛膝中脱皮甾酮的含量测定[J].中国药学杂志,2001,36(10):699-700.
[4]林大专,王广树,杨晓虹,等.牛膝中新脱皮甾酮类成分的研究[J].中国药学杂志,2006,41(17):1295-1297.
[5]孟大利,侯柏玲,汪毅.中药牛膝中的植物甾酮类成分[J].沈阳药科大学学报,2006,23(9):562-564.
[6]张翠英,李振国,张壮丽.牛膝三种酒制品脱皮甾酮的含量比较[J].中药材,2000,23(11):683-684.
[7]汪家权,赵民军,林岚.牛膝脱皮甾酮的加速溶剂提取法研究[J].中草材,2005,36(12):1797-1800.
