Generation of a Saccharomyces cerevisiae strain suitable for testing repressor function
2011-01-11LIHuiGUOQiqiang
LI Hui-e,GUO Qi-qiang
(Institute of Plateau Ecology,Tibet Agricultural and Animal Husbandry College,Nyingchi,Tibet 860000,China)
Saccharomyces cerevisiae is a simple eukaryote useful for molecular genetic studies.The best example of its utility uses transcriptional reporters to indirectly reflect the interaction between two proteins or one protein and one DNA from plant,human,animal and microbe[1-6].This strain is also used for screening activators from cDNA library.Additionally,its utility is demonstrated by the use of a yeaststrain UCC419 for testing repressor activities,where a putative repressor fused to the DNA-binding domain of GAL4 is tethered to a URA3 reporter already integrated to the host genome.If the putative repressor can repress the expression of the reporter gene URA3,it would allow for the growth of the yeast strain in the presence of FOA[7].However,especially in the case of the strain,the availability of different auxotrophic marker genes is limited.Also,the utility of cDNA library for screening activators is limited.Therefore,to expand the utility of cDNA library that was made for screening activators and UCC419 for screening repressors,we successfully re-engineered a new strain mUCC419(m:modified)that is compatible with the Invitrogen-based vector pDEST22 using PCR-based recombination strategy.
1 MATERIALS AND METHODS
The S.cerevisiae strain UCC419 was originally derived from YM725(ura3-52 his3-200 ade2-101 lys2-801trpl-901tyrl-501met-canRgal4-542 gal80-538(Mark et al.,1987)by transformation with EcoRI-SalI-digested plasmid pAadh4::GALURA3 and selection for URA+transformants[8].UCC419 was grown on-Ura media(0.67YNB,0.5%dextrose,0.069%Uracil drop out amino acid mix)to select for the URA3.For selection of UCC419m,yeast were grown on-Ura-Trp media(0.67YNB,0.5%dextrose,0.067%Uracil and Tryptophan drop out amino acid mix).
1.1 Primers and PCR
Primers 5-AGCAATATATATATATATATTTCAAGGATATACCATTCTAATGTCTGTTATTAATTTCAC(forward)and 5-CTTCTTCGGCGACAGCATCACCGACTTCGGTGGTACTGTTCTATTTCTTAGCATTTTTGa(reverse)were used to amplify TRP1 coding domain with flanking regions of LEU2.Of the 60 nucleotide(nt)length primers,40 nts of each primer are homologous to the LEU2 flank sequence(underlies),20 nts are specific to TRP1 coding region.The template for the TRP1 CDS was pDEST22(Invitrogen).The fragment was amplified with high fidelity Taq polymerase(Fermenta)with PCR protocol as described[9].The product was then size fractionated on a 1%gel in TBE buffer.The band of correct size was excised and purified.Primers 5-AGGTGGTTAGCAATCGTC(forward)and 5-TACGAAACACGCCAACCA(reverse)were used to test recombination,in which,forward primer is specific to 5’UTR of LEU2 and the reverse specific to the region of TRP1 CDS.The fragment was amplified from yeast genomic DNA by Taq polymerase(Genescript)with the following protocol:3 min at 94℃,35 cycles(30 s at 94℃,30 s at 50℃,45s at 72℃),and final extension of 10min at 72℃.
1.2 Transformation and screening
The transformation was performed according to high efficiency transformation procedure[10].About 1μg DNA of the PCR fragment was used for transformation.Transformants were selected on-Ura-Trp medium with incubation at 30℃for 3 days.Each single colony was steaked on-Ura-Trp and-Ura-Leu respectively for further testing.As For PCR screening,genomic DNAs of individual colony were prepared essentially as described[11]from 2 days cultures.PCR positive colony was cultured and then transformed with pDEST22 with transformation procedure above to restore the phenotype.
2 Results
2.1 Strategy in re-engineering UCC419
To re-engineer UCC419 so that it will be compatible with the Invitrogen-based vector pDEST22,a simple strategy was devised to convert UCC419(which is leu2+but TRP1-)to TRP1+and leu2-.The availability of the whole-genome sequence of yeast together with PCR technology allows us to precisely deletion the LEU2 gene(Illustrated in Fig.1 ).Specifically,PCR product consisting TRP1 coding sequences flanked by sequences identical to the upstream and downstream sequences of the yeast LEU2 open reading frame,respectively,was transformed into UCC419.Homologous recombina-tion in the UCC419 host involving double crossover flanking the LEU2 CDS would result in the deletion of LEU2 and replacing LEU2 with TRP1,rendering the strain leu2-and TRP1+.The TRP1 gene now is under the control of LEU2 promoter.
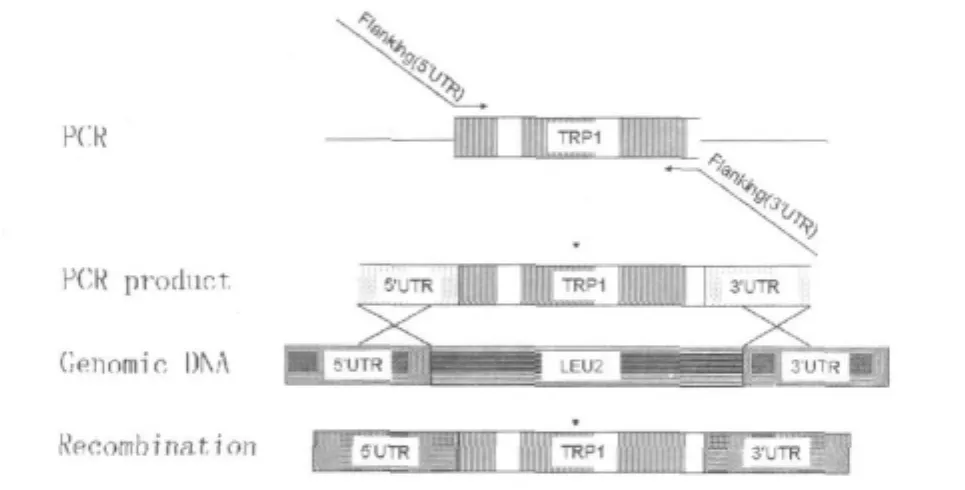
Fig.1 Schematic illustration for interested gene replaced by a marker gene flanked by homology regions
2.2 Phenotype confirming
If LEU2 is replaced by the TRP1 in UCC419,the transformants should only grow on-Ura-Trp but not-Ura-Leu media.By streaking each single colony from selective media on-Ura-Trp and-Ura-Leu media respectively,we could eliminate colonies that could grow on both-Ura-Trp and-Ura-Leu media.Because trp1 in UCC419 is a coding sequence deletion mutation[8,12].By selecting colonies that only grew on-Ura-Trp but not on-Ura-Leu media,we are able to identify recombination events at the site of LEU2 and eliminate false positive(Fig.2 ).
2.3 PCR screening
Ten colonies that could grow on-Ura-Trp media but not on-Ura-Leu media were further verified by PCR to make sure they result from recombination at the LEU2 locus.They were inoculated and grown in liquid-Ura-Trp media for 2 days,and their genomic DNA were extracted.PCR was carried out,in which the forward primer is specific to 5’UTR of LEU2 and the reverse primer is specific to TRP1 CDS.If the recombination had occurred at the LEU2 locus,causing the 5'UTR of LEU2 adjacent to the TRP1 CDS,the PCR would yield a 366bp band.Six out of 10 colonies gave rise to the expected bands,in which,#8 gave the strongest band(Fig.3 ).

Fig.2 Five colonies grew on-Ura-Trp and-Ura-Leu media respectively

Fig.3 Genomic PCR results from 10 different colonies

Fig.4 Transformants grew on-ura-leu media
2.4 Phenotype Restore
We transformed pDEST32 possessing the LEU2 gene into the newly constructed yeast UCC419m(colony#8 shown in Fig.3 )to see if it can rescue the leu2-phenotype.We also simultaneously transformed pDEST22,which does not carry LEU2,into UCC419m(colony#8)as a negative control.Transformants were selected on-Ura-Leu media.The result showed pDEST32 rescued the phenotype while pDEST22 did not(Fig.4 ).This further confirmed that UCC419m has been successfully generated.
3 CONCLUSION&DISCUSSION
The regulation of gene activity is a crucial factor in coordinating development and growth of any organism.The regulation may be either activation or repression.Yeast two hybrid system has been widely used for elucidation of activation mechanism,however,the repression one is still poorly understood because of lacking proper tools.In order to quickly engineer a yeast strain for screening or testing repressors activities,PCR-based gene replacement method was performed in this study.This method is easier and faster than the conventional plasmid-based strategy that requires restriction digestion and insertion of the marker gene into a cassette in a plasmid[13-15].The new strain mUCC419 could be used for screening repressors from our already constructed cDNA library that was for screening activators.Our work also indicates that PCR-based recombination strategies can be simply used for engineering other yeast strains for certain purpose.
Reference
[1] Degrado-Warren J,Dufford M,Chen J,et al.Shattuck D,Frech GC.Construction and characterization of a normalized yeast two-hybrid library derived from a human protein-coding clone collection[J].Biotechniques,2008,44(2):265-273.
[2] Klein P,Dietz KJ.Identification of DNA-binding proteins and protein-protein interactions by yeast one-hybrid and yeast twohybrid screen[J].Plant Stress Tolerance:Methods and Protocols,2010,639:171-192.
[3] Ngamurulert S,Limjindaporn T,Auewarakul P.Identification of cellular partners of Influenza A virus(H5N1)non-structural protein NS1 by yeast two-hybrid system[J].Acta Virologica,2009,53(3):153-159.
[4] Shen Y,Shen ZH,Zuo JK.Construction of cDNA library from mouse oocyte for yeast-2-hybrid system by homologous recombination[J].Fudan Xuebao(Yixueban),2007,34(4):599-602.
[5] Shi HX,Ren JL,Xu HZ,et al.Upregulated expression of hITF in Crohn's disease and screening of hITF interactant by a yeast two-hybrid system[J].Digestive Diseases and Sciences,2010,55(10):2929-2939.
[6] Yang JL,Zhao XM,Yin H,et al.Analysis of proteins interacted with OIPK by yeast two-hybrid method[J].Chinese Journal of Applied and Environmental Biology,2010,16(4):474-477.
[7] Zaman Z,Ansari AZ,Koh SS,et al.Interaction of a transcriptional repressor with the RNA polymerase II holoenzyme plays a crucial role in repression[J].Proceedings of the National A-cademy of Sciences,2001,98:2550-2554.
[8] Aparicio OM,Gottschling DE.Overcoming telomeric silencing:a trans-activator competes to estabhsh gene expression in a cell cycle dependent way[J].Genes&Development,1994,8:1133-1146.
[9] Brachmann CB,Davies A,Cost GJ,et al.Designer deletion strain derived from Saccharomyces cerevisiae S288C:a useful set of strains and plasmids for PCR-mediated gene disruption and other applications[J].Yeast,1998,14:115-132.
[10] Gietz,RD,Schiestl RH.High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method[J].Nature Protocols,2007,2(1):31-34.
[11] Harju S,Fedosyuk H,Peterson KR.Rapid isolation of yeast genomic DNA:Bust n’Grab[J].BMC Biotechnology,2004,4(21):4-8.
[12] Mark J,Jim D.Mutations that inactivate a yeast transcriptional regulatory protein cluster in an evolutionarily conserved DNA binding domain[J].Proceedings of the National Academy of Sciences,1987,84:2401-2405.
[13] Antoni AD,Gallwitz D.A novel multi-purpose cassette for repeated integrative epitope tagging of genes in Saccharomyces cerevisiae[J].Gene,2000,246:179-185.
[14] Zaragoza O.Generation of disruption cassettes in vivo using a PCR product and Saccharomyces cerevisiae[J].Journal of Microbiological Methods,2003,52:141-145.
[15] Walker M,Vystavelova A,Pedler S,et al.PCR-based gene disruption and recombinatory marker excision to produce modified industrial Saccharomyces cerevisiae without added sequence[J].Journal of Microbiological Methods,2005,63:193-204.
