顺序注射催化光度分析法测定环境水样中痕量钼
2011-01-11王夕云陈志霞范世华
王夕云 陈志霞 范世华
(东北大学分析科学研究中心,辽宁沈阳 110004)
顺序注射催化光度分析法测定环境水样中痕量钼
王夕云 陈志霞 范世华*
(东北大学分析科学研究中心,辽宁沈阳 110004)
基于钼(V I)对碘化钾/过氧化氢反应体系催化作用的反应产物在350 nm处的吸光度与钼在一定浓度范围内呈线性关系的原理,建立了灵敏的顺序注射催化动力学光度分析新方法。方法的线性范围是2~300μg/L,方法的检出限(3σ)为1.1μg/L,相对标准偏差(RSD)为0.6%(n=11)。每次测定仅消耗100μL试样,75μLKI和125μLH2O2,对4个不同水样的钼测定回收率在95.2%~105.2%之间。顺序注射催化动力学光度分析测定环境水样中痕量钼的方法是一种快速、准确的新方法。
顺序注射;催化光度法;钼
1 引言
钼是元素周期表中第二过渡系元素中唯一具有特殊生物功能的元素,广泛存在于土壤,水,空气,动植物组织中。钼是一切固氮植物所必需的营养成分,也是人体必需的微量元素,主要通过食物链进入人体。钼是三种不同酶系统―黄嘌呤氧化酶、黄嘌呤脱氢酶和醛氧化酶的成分。钼的生理功能是通过各种含钼酶的活性来实现的。对哺乳动物的遗传物质有保护作用;缺钼可导致儿童和青少年发育不良,智力发育迟缓,影响骨骼生长,而且可能会引起急性心肌炎,肾结石,大骨节和食道癌等疾病,且易患高血压,糖尿病。近年来的研究表明,钼是一种抗癌元素,适量钼的存在可以降低致癌物质的毒性,由于钼具有特殊的生理生化功能,在生命科学研究领域日益引起关注。
已报道的钼的测定方法有火焰原子吸收光谱法[1]和石墨炉原子吸收光谱法[2-4],电感耦合等离子体原子发射光谱法[5-6],ICP-MS法[7-9],电化学法[10-12]等。
光度分析方法因其设备简单,价格低廉,易于普及等特点也常用于钼的测定。新的显色剂和显色体系的发现,多波长光度法的发展与应用,使分光光度法测定钼的分析灵敏度和选择性有较大的提高 [13-15]。
将顺序注射进样与分光光度法联用,利用酸性介质中钼(V I)催化 KI和H2O2的氧化还原反应产生的I2,溶于KI形成KI3后吸光度的变化与钼的浓度在一定范围内成线性响应关系的原理,建立了测定痕量钼的新方法,快速、准确地测定了环境水样中钼的含量。
2 实验部分
2.1 实验仪器
722光栅分光光度计(上海第三分析仪器厂); KLOEHN顺序注射进样系统(KLOEHN,USA); PCL-711B数据采集卡(Advantech,台湾);CQ-6超声波清洗器(上海沪超超声波仪器有限公司); 90005-02纯水系统(LABCONCO)。
2.2 试剂及配制
碘化钾、过氧化氢、钼酸铵、盐酸均为AR级。
所用水均为二次去离子水。
钼酸铵标准储备液(100 mg/L,以钼浓度表示):称取0.0920 g(NH4)6Mo7O24·4H2O到小烧杯中,加水溶解后转移至500 mL的容量瓶中,加水定容至刻度线,摇匀。用时根据需要稀释成不同浓度的标准溶液。
碘化钾储备液(10%):称取10.0 g KI到烧杯中,加水溶解后转移到100 mL容量瓶中,加水定容至刻度线,摇匀。根据实验需要配制成不同浓度的工作溶液。
过氧化氢(3%):移取10.0 mL30%的 H2O2,稀释定容到100 mL的容量瓶中,摇匀。用时,稀释至适当的浓度。
盐酸储备溶液浓度为1 mol/L,用时稀释至所需的浓度。
2.3 顺序注射系统的操作过程
实验流程如图1。SI系统通过六位选择阀的一个阀位通道对整个流路进行冲洗后,依次吸入一定体积的试样、KI溶液和 H2O2溶液(含一定浓度HCl)进入储存管路进行混合,然后改变泵的运行方向,再将此混合溶液输送至流通池,由检测器实时记录吸光度信号,在波长350 nm处测定溶液的吸光度。整个过程在计算机控制下按一定程序完成。
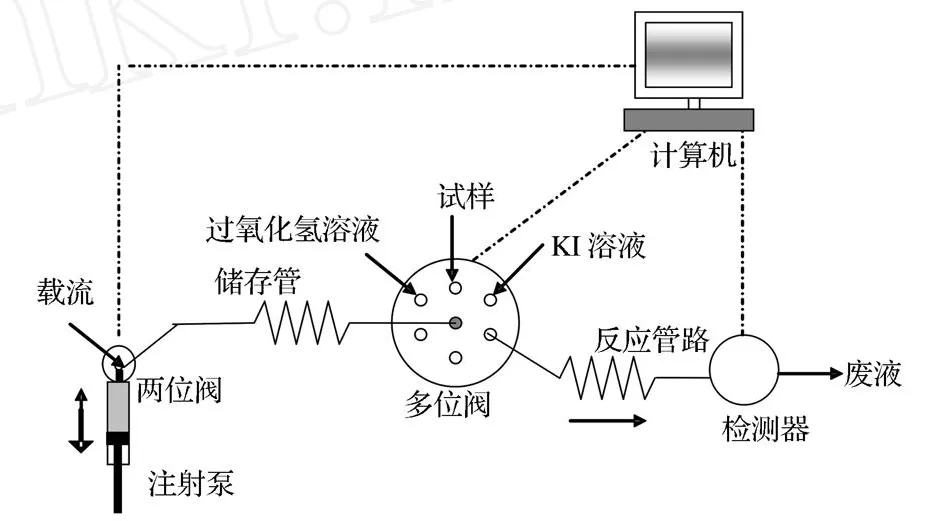
图1 顺序注射分光光度法测定钼的流程图Figure 1.Schematic diagram of the SIA system for determ ination of Mo.
3 结果与讨论
3.1 最大吸收波长
在310~510 nm的范围内,扫描了在盐酸介质中,KI与 H2O2在(NH4)6Mo7O24催化作用下氧化还原反应产物的吸收光谱,反应产物在350 nm处的吸光度最大,实验选择350 nm为最大吸收波长。
3.2 进样顺序的影响
实验中采用三区带进样方式,不同的区带排列顺序使试剂和试样的混合与反应情况不同,从而会影响反应信号的大小。分别注入100μLKI溶液(2%),100μLH2O2(0.03%)和 HCl(0.2 mol/L)的混合溶液,150μL(NH4)6Mo7O24溶液(50μg/mL),在检测流速为1.2 mL/min的条件下,通过改变试样和试剂的吸入顺序,分别测定吸光度。实验结果表明,采用“(NH4)6Mo7O24→KI→H2O2/HCl”的进样顺序得到的吸收信号最大,约为其它进样顺序的吸收信号2倍左右。这可能是因为在此种进样方式下,试样溶液最先进入储存管,增加了试样在管路中的停留时间,物理分散程度比较大,KI溶液和过氧化氢在钼的催化下发生混合渗透的反应比较充分,因而产生的吸光度信号值比较大。故实验采用“(NH4)6Mo7O24→KI→H2O2”进样顺序。
3.3 流速的影响
流速对吸光度差值的影响如图2所示。从图2可以看出,吸光度差值随流速的增大而迅速降低。其实质是流速影响到试样与试剂在反应管道内的留存时间和反应进行的时间,流速越快,试样和试剂的混合反应程度越小,反应时间越短,相对吸光度也就越低。兼顾灵敏度和分析通量,实验选定流速V为1.2 mL/min。

图2 流速V对吸光度差值的影响Figure 2.Influence of flow rate V on absorbance difference(ΔA).
3.4 试剂浓度的影响
(1)盐酸浓度
在实验中,盐酸为氧化还原反应的进行提供了一个酸性环境,所以需要对盐酸的浓度进行优化,选择一个合适的酸度条件。实验结果表明,随着盐酸浓度的增加,吸光度差值也随之迅速增加,当 HCl浓度达到0.2 mol/L后,吸光度值基本不再发生变化。因此,实验选择0.2 mol/L作为 HCl溶液的最佳工作浓度。
(2)碘化钾浓度
实验结果表明,在0.5%~2%的浓度范围内随着 KI溶液浓度的增加,吸光度逐渐增强,而在2%~6%的浓度范围内吸收信号随浓度的变化幅度逐渐减小;且 KI浓度达到6%时,绝对吸光度值已经接近2.000,所以选择 KI溶液的浓度为3%进行后续的实验。
(3)H2O2浓度
H2O2作为氧化剂,随着 H2O2浓度的增加,相对吸光度也随之增加,当 H2O2浓度达到0.03%时,吸光度差值基本不再变化,故实验选择0.03%作为H2O2的最佳浓度。
3.5 KI体积的影响
由图3可见,随着 KI体积的增加,吸光度差值也随之增加;当KI体积达到75μL时吸光度差值达到最大;继续增加 KI体积,吸光度差值则降低,这可能是因为过多的 KI试剂在反应管道中无法与试样进行充分有效的渗透混合反应,反而阻碍了第一区带与第三区带的扩散混合,导致信号值降低。因此,实验选择75μL作为 KI的最佳工作体积。

图3 KI体积对吸光度差值的影响Figure 3.Influence of KIvolume on absorbance difference(ΔA).
3.6 试样体积的影响
试样体积影响到参与反应物质的总量以及试剂和试样反应的完全程度,从而影响测定灵敏度。试样体积对吸光度差值的影响如图4所示。

图4 试样进样体积对吸光度差值的影响Figure 4.Influence of sample volume on absorbance difference(ΔA).
从图中可看出,随着试样体积的增加,相对吸光度值也随之增加,当试样体积达到100μL后再继续增加体积时,吸光度值基本不再变化,故实验选择100μL作为试样的最佳进样体积。
3.7 H2 O2体积的影响
由图5可见,随着H2O2体积的增加,吸光度差值也随之增加;当 H2O2/HCl体积达到125μL时吸光度差值达到最大;继续增加 H2O2/HCl体积,吸光度差值则降低,因此实验选择125μL作为H2O2的最佳工作体积。

图5 H2 O2进样体积对吸光度差值的影响Figure 5.Influence of H2 O2 volume on absorbance difference(ΔA).
3.8 共存离子干扰及消除
在测定结果允许误差不超过±5%的范围内,对50μg/L的Mo(V I)进行测定,实验结果表明,7500倍NO3-,1500倍的 K+、Na+、Ca2+、Mg2+,500倍的CH3COO-,PO43-,200倍的 A l3+、SiO32-,10倍的Fe2+和1倍的 Fe3+不干扰测定。对可能产生干扰的金属阳离子,用EDTA加以掩蔽。
3.9 分析性能
3.9.1 校正曲线和线性范围
在上述优化的实验条件下,即进样顺序为先吸入样品溶液,再吸入 KI溶液,最后吸入 H2O2/HCl氧化剂;进样体积分别为样品100μL,KI为75μL, H2O2为125μL;检测流速为1.2 mL/min,以吸光度差值对Mo(V I)的浓度进行线性拟合,校正曲线如图6所示。
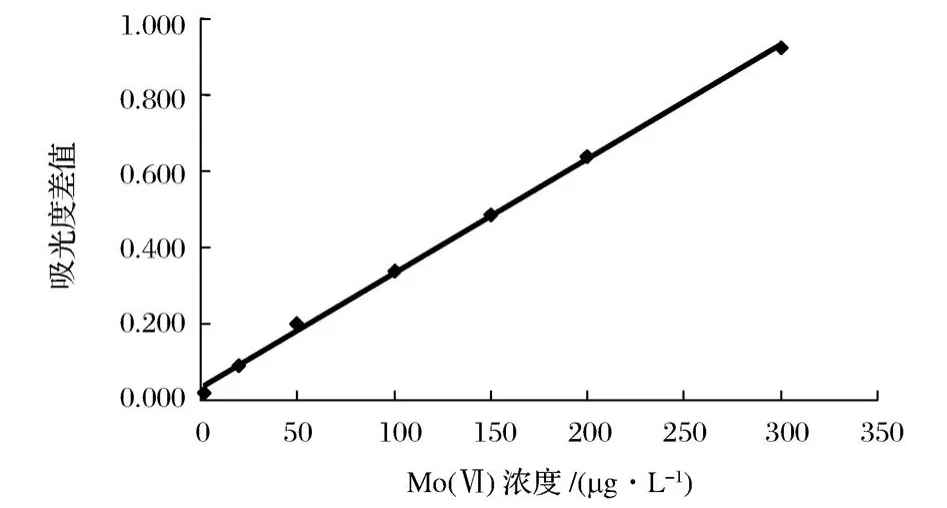
图6 Mo(VI)标准曲线Figure 6.The calibration curve for Mo(VI).
在2.0~300.0μg/L浓度范围内吸光度差值与钼浓度呈线性关系。线性回归方程为:ΔA= 0.00420c+0.0332(r=0.9992,n=7)。
3.9.2 精密度和检出限
在最优条件下,对50μg/L的试样进行连续11次平行测定,所得的相对标准偏差为0.6%(n= 11);以二次去离子水作为空白溶液,代替标准试样溶液11次平行测定,以测定空白信号吸光度的3倍标准偏差所对应的浓度作为检出限(3σ),计算得出方法的检出限(DL)为1.2μg/L。
4 实际样品的测定
分别取河水、泉水、湖水、自来水样,过滤后按2.3节进行测定。方法的回收率在 95.2%~105.2%之间(表1)。
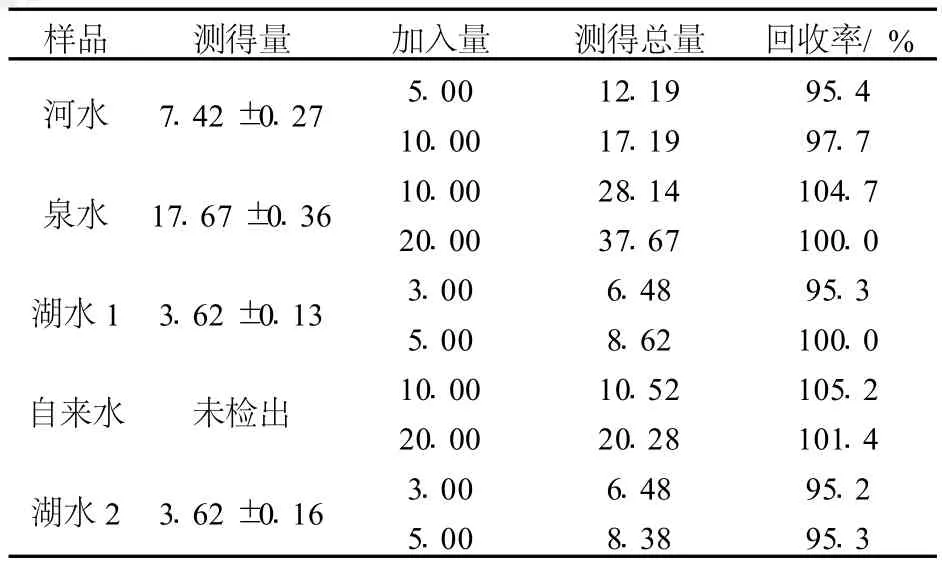
表1 实际水样测定结果(n=3)Table 1 The analytical results of the real water samples μg/L
5 结语
本文将顺序注射进样技术与分光光度法结合,建立了顺序注射催化动力学光度分析测定痕量钼的新方法。方法具有灵敏度高、分析速度快、试剂消耗少、自动化程度高的优点,是一个环境友好型的分析方法,有望用于环境水样中痕量钼的批量测定。
[1]Yigmatepe E,Yaman M.Determination of molybdenum in biological samples by flame atomic spectrometry after p reconcentration on activated carbon[J].Monatshefte fur chemie,2011,142(2):131-136.
[2]Shamsipur M,Habibollahi S.A highly sensitive p rocedure for determination of ultra trace amounts of molybdenum by graphite furnace atomic abso rp tion spectrometry after dispersive liquid-liquid microextraction[J].Microchim.Acta,2010,171(3-4):267-273.
[3]Filik H,Cengel T,Apak R.Selective cloud point extraction and graphite furnace atomic absorption spectrometric determination of molybdenum(V I)ion in seawater samples[J]. J.Hazard.Mater.,2009,69(1-3):766-771.
[4]Liang P,Li Q,Liu R.Determination of trace molybdenum in biological and water samp les by graphite furnace atomic absorp tion spectrometry after separation and p reconcentration on immobilized titanium dioxide nanoparticles[J].Microchim.Acta,2009,164(1-2):119-124.
[5]杜米芳.电感耦合等离子体发射光谱法快速测定钼铁合金中的钼[J].岩矿测试,2010,29(1):89-90.
[6]Kalal H S,Panahi H A,Framarzi N,et al.New chelating resin for p reconcentration and determination of molybdenum by inductive coup le p lasma atomic emission spectroscopy[J].Int.J.Environ.Sci.Technol.,2011,8(3): 501-512.
[7]Pearce C R,Cohen A S,Parkinson IJ.Quantitative Separation of Molybdenum and Rhenium from Geological Materials for Isotopic Determination by MC-ICP-MS [J].Geostand.Geoanal.Res.,2009,33(2):219-229.
[8]Reid H J,Basharnmakh A A,Goodall PS,et al.Determination of iodine and molybdenum in milk by quadrupole ICP-MS[J].Talanta,2008,75(1):189-197.
[9]冯先进,屈太原.电感耦合等离子体质谱法(ICP-MS)最新应用进展[J].中国无机分析化学,2011,1(1):46-52.
[10]Nakiboglu N,Tunay Z,Sahin I.Determination of molybdenum by adso rp tive catalytic stripping voltammetry in thee p resenceof alizarin reds and persulphate[J].Fresenius Environ.Bull.,2011,20(4):939-944.
[11]Neelam Y S,Dasari R,Prasad P R,et al.Determination of molybdenum(V I)by differential pulse polarographic technique using 4-(2-hydroxy phenyl ethaminodiol) benzene-1,3-diol(4-2-HPEDB-1,3,D)[J].J.Saudi Chem.Soc.,2010,14(2):149-155.
[12]Deng P H,Fei J J,Feng Y L.Trace determination of molybdenum by anodic adso rp tive stripping voltammetry using amulti-walled carbon nano tubesmodified carbon paste electrode[J].Sens.Actuators:B,2010,148 (1):214-220.
[13]Madrakian T,Afkhami A,Siri R,et al.Micellemediated extraction and simultaneous spectrophotometric determination of vanadium(V)and molybdenum(V I)in plant foodstuff samp les[J].Food Chem.,2011,127 (2):769-773.
[14]Kumar A,Dass R.A rapid spectrophotometric method for the determination of molybdenum using ferron in industrial and environmental samp les[J].Indian J. Chem.,Sect A,2008,47(10):1533-1536.
[15]Madrakian T,Ghazizadeh F.Cloud-point p reconcentration and spectropho tometric determination of trace amounts of molybdenum(V I)in steels and water samples[J].J.Hazard.Mater.,2008,153(1-2):695-700.
Determ ination of Molybdenum atμg/LLevels in Environmental Water Samples by Catalytic Spectrophotometric Sequential Injection Analysis
WANG Xiyun,CHENZhixia,FANShihua*
(Research Center for Analytical Science,Northeastern University,Shenyang,Liaoning110004,China)
On the basisof a linear relationship between Mo(V I)concentration and the absorbance(at 350 nm)of KI3,the p roductof the reaction of potassium iodide(KI)with hydrogen peroxide(H2O2)using Mo(V I)as the catalyst,a sensitive catalytic spectrophotometric sequential injection analysismethod was developed for the determination of Mo(V I)atμg/Llevels.The calibration curve is linear over the range of 2~300μg/Lwith a detection limit(3σ)of 1.1μg/L.The relative standard deviation(RSD)is 0.60%for 50μg/Lmolybdenum(n=11). Molybdenum w as determ ined in environmental w ater samp les at a samp ling rate of 80 h-1with 100μLsample,75μLKIand 125μLH2O2reagent injections.The recoveries of the method tested by 4 different w ater samp les are 95.2%~105.2%.It concludes that the method is a new app roach to p rovide a fast and accurate analysis of trace amount of molybdenum in environmental water samp les.
sequential injection;catalytic spectrophotometry;molybdenum
O657.32;TH744.12+2
A
2095-1035(2011)03-0027-05
10.3969/j.issn.2095-1035.2011.03.0005
2011-07-08
2011-07-28
辽宁省高校重点实验室资助项目(2008S093)。
王夕云,女,硕士,流动分析技术研究。
范世华,男,教授,流动分析技术研究。E-mail:shfneu@sina.com
