双羟基脲与Fe(Ⅲ)的络合及氧化还原反应
2011-01-09晏太红张柏青郑卫芳卞晓艳李传博
晏太红,张柏青,郑卫芳,张 虎,左 臣,张 宇,鲜 亮,卞晓艳,李传博
中国原子能科学研究院放射化学研究所,北京 102413
双羟基脲与Fe(Ⅲ)的络合及氧化还原反应
晏太红,张柏青,郑卫芳,张 虎,左 臣,张 宇,鲜 亮,卞晓艳,李传博
中国原子能科学研究院放射化学研究所,北京 102413
用分光光度法和循环伏安分析法研究了双羟基脲(DHU)与Fe(Ⅲ)的作用,结果表明,DHU与 Fe3+能形成紫色配合物,配合物有二级或以上络合,形成的1∶1配合物分解速率对配合物来说为1级,在10℃下,c0(DHU)=c0(FeCl3)=1.0×10-3mol/L时,配合物表观一级分解速率常数k′=0.031 min-1。其配位方式可能是其分子中的N—O上的氧原子和另一个N—O上的氮原子与 Fe3+配位。但形成的配合物不稳定,会发生配合物分子内氧化还原反应,电子从DHU分子转移到 Fe(Ⅲ),Fe(Ⅲ)被还原为 Fe(Ⅱ),而DHU被氧化。
双羟基脲;Fe(Ⅲ);配位;氧化还原;反应动力学
双羟基脲(DHU)作为一种新型有机还原剂,能快速还原 Pu(Ⅳ)和Np(Ⅵ),在 Purex流程铀钚分离中具有潜在的应用前景[1-3]。同时其分子结构(HOHNCON HOH)与羟肟酸、羟基脲类似,具有—CONHOH基团,其络合性质可能与羟肟酸、羟基脲相似,对一些锕系元素离子具有一定的络合能力[4-6],早在20世纪60—70年代,Boyland等[7-8]报道了DHU的合成及作为药物其在诱导染色体变异方面的初步研究。但目前尚未见关于DHU与金属离子的络合性质的公开报道,人们对其络合及其氧化还原性质的了解非常有限。Harmon等[9]报道了羟基脲(HU)及乙基羟基脲与金属离子如 Fe(Ⅲ)等的配位行为,研究表明,羟基脲及乙基羟基脲首先与Fe(Ⅲ)形成1∶1的配合物,配合物在560 nm处有吸收。乙基羟基脲与Fe(Ⅲ)形成的配合物在水溶液中不稳定,迅速分解放出N2O、CO2等气体,配合物分解涉及分子内氧化还原反应。近年来关于羟基脲与 Fe(Ⅲ)的配位及氧化还原机理研究较多,Bedrica等[10]研究了高氯酸体系中 HU与Fe(Ⅲ)的配位行为,发现 HU对Fe(Ⅲ)的络合能力要低于羟肟酸的络合能力。Nigovic’等[11]采用循环伏安法和电子顺磁共振(EPR)技术研究了羟基脲与Fe(Ⅲ)配合物氧化还原反应中的电子转移机理,配合物分子内配体单电子转移到Fe(Ⅲ),Fe(Ⅲ)被还原到Fe(Ⅱ),EPR谱显示反应过程中有自由基H2N—CO—NHO·产生。但到目前为止尚未见DHU与Fe(Ⅲ)反应的报道。本工作拟通过分光光度法和循环伏安法系统研究DHU与FeCl3的络合及氧化还原作用,以期获取DHU与金属离子的络合及氧化还原反应数据,深入认识DHU的络合及氧化还原性质,为其在锕系元素分离等方面的实际应用提供指导。
1 实验部分
1.1 主要仪器与试剂
Specord600型二极管阵列分光光度计,德国Jena公司产品;CHI660C电化学工作站,上海辰华仪器有限公司产品,工作电极为CHI102型铂电极,辅助电极为CHI115型铂丝电极,参比电极为饱和甘汞电极;5973型气相色谱-质谱联用仪,美国Agilent公司产品。
双羟基脲(DHU),本实验室合成,纯度大于97%;FeCl3·6H2O,分析纯,广东汕头西陇化工厂产品;其余试剂均为分析纯。
1.2 实验方法
1.2.1 分光光度法研究双羟基脲与Fe(Ⅲ)的反应 分别将2.0×10-4mol/L和1.0×10-3mol/L FeCl3水溶液加入石英比色池中,然后分别加入一定量的DHU水溶液,用分光光度计监测溶液吸收光谱的变化。同样方法研究水-乙醇体系中该反应。反应后气态产物采用气相色谱-质谱联用仪进行检测。
1.2.2 电化学方法研究双羟基脲与Fe(Ⅲ)的反应 以0.1 mol/L KCl作支持电解质,向5.0×10-3mol/L FeCl3中加入不同量的DHU,然后在电化学工作站用循环伏安法测量溶液的循环伏安曲线。甘汞电极作参比电极,铂电极作工作电极,扫描速率为100 mV/s。
2 结果与讨论
2.1 分光光度法研究双羟基脲对Fe(Ⅲ)的配位及还原
与其它羟肟酸一样,室温(20℃)下,向2.0×10-4mol/L FeCl3水溶液中加入1.2×10-4mol/L DHU水溶液后,溶液立即呈深紫色,这是由于DHU分子结构中含有—C=ONHOH基团,它与Fe(Ⅲ)形成紫色配合物,但随后溶液深紫色逐渐褪去。反应过程中溶液吸收光谱变化示于图1。由图1可以看出,FeCl3水溶液在300 nm处有吸收,该峰可以归属为电荷迁移跃迁带。加入DHU后,300 nm处的吸收峰迅速变弱,在360 nm处的较弱吸收峰显现,而在525 nm左右出现宽的吸收峰,该峰可以归属为d-d跃迁谱带,电子从中心原子的d轨道跃迁到较高能级的d轨道,这种跃迁是对称性禁阻的,电子运动与振动的耦合使得这种禁阻得以暂时解除,但强度较小。与羟基脲相比,其 d-d跃迁向短波移,这可能是因为DHU产生的配位场较羟基脲强,当水被配位较强的配体取代时,d-d轨道间能级差变大,导致d-d跃迁向短波移。同样在水-乙醇体系中也可以看出类似的变化(图2)。通过高斯多峰模拟可以更清楚地看到300~400 nm范围内其实有300 nm、360 nm两个吸收峰。

图1 FeCl3水溶液中加入DHU后吸收光谱的变化Fig.1 Variation of UV-Vis spectra of FeCl3 solution after addition of DHU

图2 FeCl3水-乙醇溶液中加入DHU后吸收光谱的变化Fig.2 Variation of UV-Vis spectra of FeCl3in water-ethanol solution after addition of DHU
由图1、2还可看出,525 nm处出现的吸收峰又很快消失,这说明形成的配合物又快速分解,与羟基脲一样,这可能是由于络合后配合物中配体的电子转移到 Fe(Ⅲ)上而发生氧化还原反应。反应完成后用气相色谱-质谱联用仪检测反应产物。图3为气态产物气相色谱分离后的质谱图。由图3可看出,质谱检测出了N2O的分子离子峰,这表明气态产物中有N2O。
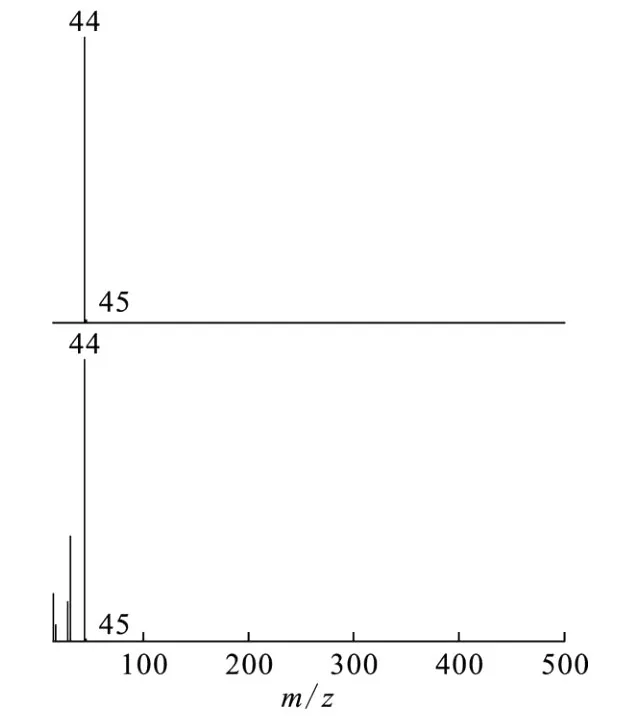
图3 气态产物经气相色谱分离后N2O的质谱图Fig.3 Mass spectra of N2O separated from the gas products by gas chromatography
由于在525 nm左右的吸收峰较弱,反应不易监测,另外20℃下该反应进行较快,不易检测反应初始过程,因此实验时提高 FeCl3初始浓度为1.0×10-3mol/L,然后分别加入不同量的DHU,在10℃下考察反应液在525 nm处吸光度随时间的变化情况,示于图4(a)。由图4(a)可看出,随着DHU浓度的增加,配合物形成速率加快,c0(DHU)=5.0×10-4mol/L时,吸光度在反应3 min后达到最大,c0(DHU)=1.0×10-3mol/L时,吸光度在反应几秒钟时达到最大。而当c0(DHU)/c0(FeCl3)>1时,仪器就监测不到吸光度的最大值;随着DHU浓度的增加,形成的配合物分解速度也加快。c0(DHU)/c0(FeCl3)≤1时,配合物形成后数分钟内吸光度未见下降。c0(DHU)/c0(FeCl3)>1时,随着DHU浓度的增加,吸光度迅速下降,配合物分解速度加快。

图4 c0(FeCl3)=1.0×10-3mol/L时不同DHU初始浓度时溶液在525 nm处吸光度随时间的变化Fig.4 Variation of absorbance at 525 nm with time at different initial concentration of DHU and c0(FeCl3)=1.0×10-3mol/L
以lnA对时间作图示于图4(b)。由图4(b)可见,c0(DHU)/c0(FeCl3)≤2时,配合物形成后lnA对时间为线性关系,而当c0(DHU)/c0(FeCl3)更大时则偏离线性关系,推测DHU与FeCl3首先形成1∶1的配合物,该配合物分解速率对配合物来说为1级,而当c0(DHU)更大时,则会形成2∶1或更高的配合物,这种配合物在525 nm左右也有吸收峰,且这种配合物分解速度更快,那么这种情况下的吸光度A表观=ε1bc(Fe·DHU2+)+ε2bc(Fe·2DHU+),其中b为液池厚度,Fe·DHU2+和 Fe·2DHU+同时分解,而Fe·2DHU+分解又会产生 Fe·DHU2+,因此lnA表观对时间偏离线性关系。
以c0(DHU)/c0(FeCl3)对吸光度A作图(图 5),在c0(DHU)/c0(FeCl3) ≤1 时 ,据A=εbc可求得1∶1配合物摩尔消光系数ε约为611.5 L/(mol·cm),但当c0(DHU)/c0(FeCl3)继续增大时吸光度继续呈线性增大,两条直线相交对应首先形成1∶1的配合物,然后又形成2∶1的配合物。
进一步提高溶液 FeCl3初始浓度为2.5×10-3mol/L,考察c0(DHU)/c0(FeCl3)较低时的情况,得到反应溶液在525 nm处吸光度随时间的变化情况示于图6。与前述结果一致,c0(DHU)/c0(FeCl3)≤1时,主要形成1∶1的配合物,配合物形成后 lnA对时间为线性关系,而当c0(DHU)/c0(FeCl3)更大时则偏离线性关系。
前面提到,形成的1∶1配合物分解速率对配合物来说为1级,这也可以从下面的分析得到印证,当c0(DHU)/c0(FeCl3)≤1时,配合物形成反应可以表示为:

图5 吸光度A与c0(DHU)/c0(FeCl3)的关系Fig.5 Relationship between absorbance and c0(DHU)/c0(FeCl3)
假定该反应进行得较完全,那么c0(DHU)=c(FeCl3·DHU),对于配合物分解反应:

以图6(b)中配合物形成后得到的直线斜率与lnc0(DHU)作图,示于图7,线性模拟得一直线斜率为1.1,在实验误差范围内可以认为为1,这同样说明分解反应对于配合物来说为1级。
对于一级反应来说,其速率方程可以写为:


图6 c0(FeCl3)=2.5×10-3mol/L时不同DHU初始浓度时溶液在525 nm处吸光度随时间的变化Fig.6 Variation of absorbance at 525 nm with time at different initial concentration of DHU and c0(FeCl3)=2.5×10-3mol/L

图 7 lnk′与 lnc0(DHU)的关系Fig.7 Relationship between lnk′and lnc0(DHU)
其中x为分解掉的配合物的浓度。但显然分解反应并不属于简单的一级反应,还有其它副反应发生。因此只能得到表观一级分解速率常数,在c0(DHU)=c0(FeCl3)=1.0×10-3mol/L时,k′=0.031 min-1。
由于该反应是连续反应,且还涉及到Fe3+的水解及其它副反应,因此要得到其真实的反应机理,尚需作进一步深入的研究。
2.2 循环伏安法分析
采用循环伏安法进一步研究了DHU与FeCl3的配位以及配合物的电子转移机理。图8为FeCl3及DHU配合物的循环伏安曲线。由图8可以看出,DHU有一较强的氧化峰。Ⅰc/Ⅰa峰为Fe3+/Fe2+的准可逆峰,阳极阴极峰电势相差85 mV,这表明此为扩散控制的单电子电荷转移过程[10]。当 DHU与 FeCl3按 1∶1混合后,Ⅰc/Ⅰa峰位变化不大,阴极支 Ⅰc峰位略向负移,但 Ⅰa峰位并没有变化,这说明 DHU与FeCl3配位较弱。与羟基脲和Fe3+配位的循环伏安曲线类似,出现两个阴极峰Ⅱc、Ⅲc,这可归属于DHU氧化产物的还原峰。
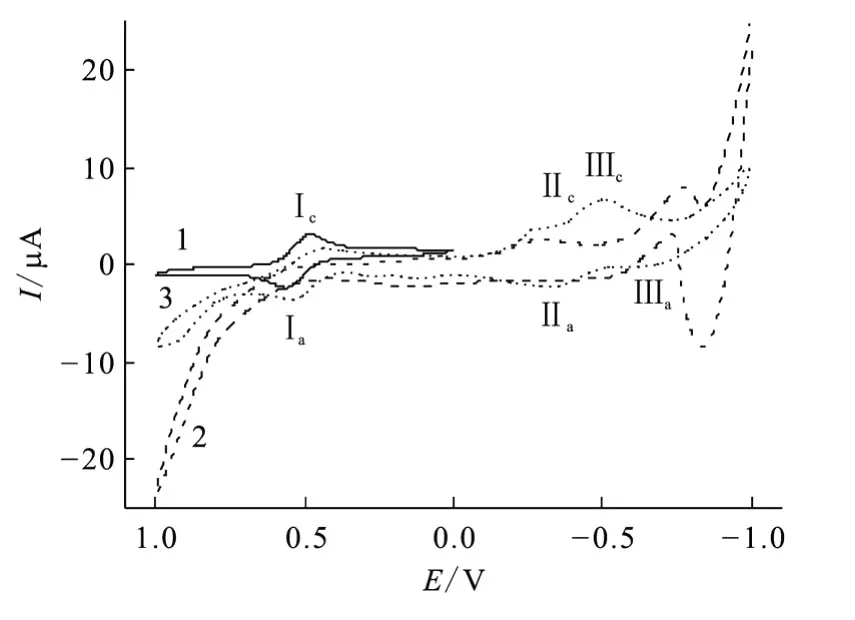
图8 FeCl3、DHU及DHU配合物的循环伏安曲线Fig.8 Cyclic voltammograms of free Fe(Ⅲ)species,dihydroxyurea,its complex with Fe(Ⅲ)
进一步改变溶液中DHU与FeCl3的比例,结果示于图9,c0(DHU)/c0(FeCl3)为0.5和1时基本没有区别,而当DHU浓度更高时(c0(DHU)/c0(FeCl3)=4),Ⅰc变弱而出现 Ⅰ′c,这可归属为与DHU配位的 Fe3+的还原峰,Ⅰ′c比Ⅰc偏负约130 mV,而羟基脲相应峰偏负约100 mV左右,这说明DHU对 Fe3+的配位比羟基脲更强一些。

图9 DHU配合物的循环伏安曲线Fig.9 Cyclic voltammograms of complex with Fe(Ⅲ)for differentc0(DHU)/c0(Fe)ratio
乙羟肟酸与Fe3+配合物的电化学行为表明配合物还原电势更负,有较高的稳定常数,说明其与Fe3+配位较双羟基脲更强[12]。由于配合物键合方式对氧化还原反应的影响要大于配体取代基效应的影响[13]。对于乙羟肟酸而言,其与Fe3+配位较强,其分子中的 2个氧原子与 Fe3+配位(图10(a)),而DHU 配位则不同(图 10(b)),其配位方式可能是其分子中的N—O上的氧原子和另一个N—O上的氮原子与Fe3+配位。
对比这些结果可知,在DHU与FeCl3间发生了氧化还原反应,电子从DHU转移到 Fe3+,Fe3+被还原为 Fe2+,而DHU给出电子,被氧化为 N2O、CO2等产物。

图10 乙羟肟酸(a)和DHU(b)与Fe3+可能的配位方式Fig.10 Possible schematic structure of the complex of Fe3+ion with acetohydroxamic acid(a)and DHU(b)
3 结 论
用分光光度法和电化学分析方法研究的结果表明,DHU与Fe3+能形成紫色配合物。配合物可能有二级或以上配位,其配位方式可能是其分子中的N—O上的氧原子和另一个N—O上的氮原子与Fe3+配位。热力学分析表明形成的配合物不稳定,会发生配合物分子内氧化还原反应,电子从DHU转移到 Fe3+,Fe3+被还原为 Fe2+,而DHU给出2个电子,被氧化为N2O等产物。由于该反应是连续反应,且还涉及到 Fe3+的水解及其它副反应,因此要得到其真实的反应机理,尚需作进一步的深入研究。
[1]Yan Taihong,Zheng Weifang,Zuo Chen,et al.The Reduction of Np(Ⅵ)and Np(Ⅴ)by Dihydroxyurea and Its Application to the U/Np Separation in the PUREX Process[J].Radiochim Acta,2010,98(1):35-38.
[2]Yan Tai-hong,Zuo Chen,Zheng Weifang,et al.Kinetics of Reductive Stripping of Pu(Ⅳ)in the TBP/OK-HNO3System Using Dihydroxyurea[J].J Radioanal Nucl Chem,2009,280(3):585-588.
[3]Yan Taihong,Zheng Weifang,Ye Guo-an,et al.Synthesis of Dihydroxyurea and Its Application to the U/Pu Split in the PUREX Process[J].J Radioanal Nucl Chem,2009,279(1):293-299.
[4]May I,T aylor R,Denniss J I S,et al.Neptunium(Ⅳ)and Uranium(Ⅵ)Complexation by Hydroxamic Acids[J].J Alloys Comps,1998,275 277:769-772.
[5]Sinkov S I,Choppin G R,Taylor R J.Spectrophotometry and Luminescence Spectroscopy of Acetohydroxamate Complexes ofTrivalent Lanthanide and Actinide Ions[J].J Solution Chem,2007,36:815-830.
[6]朱兆武.羟基脲——新型无盐试剂在 Purex流程中的应用研究[D].北京:中国原子能科学研究院,2003.
[7]Boyland E,Nery R.The Synthesis and Some Reactions of Dihydroxyurea[J].J Chem Soc(C),1966:350-353.
[8]Boyland E,Nery R.Biochemistry Dihydroxyurea[J].Nature,1964,26(4 952):1 379-1 380.
[9]Harmon R E,Dabrowiak J C,Brown D J,et al.Metal Complexes of 1-Substituted 3-Hydroxyureas[J].J Med Chem,1970,13(3):577-579.
[10]Bedrica A,Birus M,Kujundzic N.Iron(Ⅲ)Complexation by Hydroxyurea in Acidic Aqueous Perchlorate Solution[J].Croatica Chem,1988,61(1):21-31.
[12]Escot M T,Pouillen P,Martinet P.Etude,par voie Electrochinque,de l’Influence du fer(Ⅱ)sue la Reduction de Cetones en Solution dans leN,NDimethylformamide[J].Electrochim Acta,1983,28(12):1 697-1 702.
[13]Nigovic’B,Kujundž žic’N.Electrochimical Behavior ofIron(Ⅲ) ComplexesWith Aminohydroxamic Acids[J].Polyhedron,2002,21(16):1 661-1 666.
Coordination and Redox Reaction of Dihydroxyurea With Fe(Ⅲ)
YAN Tai-hong,ZHANGBai-qing,ZHENG Wei-fang,ZHANG Hu,ZUO Chen,ZHANG Yu,XIAN Liang,BIAN Xiao-yan,LI Chuan-bo
China Institute of Atomic Energy,P.O.Box 275(26),Beijing 102413,China
The interaction of dihydroxyurea(DHU)with Fe(Ⅲ)was studied by spectrophotometry and cyclic voltammetry.The results show that DHU forms dark purple complex quickly after mixing with Fe(Ⅲ).Whereas the complex formed is relatively unstable.The decomposition rate constant of the complexk′is estimated to be 0.031 min-1at 10 ℃andc0(DHU)=c0(FeCl3)=1.0×10-3mol/L.Chelation through the N—O oxygen atom and N—O nitrogen atom can be assumed for the complex.It is found that the innermolecular redox occurred between DHU and Fe(Ⅲ)after the formation of complex in solution.The one-electron transfer from DHU to Fe(Ⅲ)is assumed.Fe(Ⅲ)is reduced to Fe(Ⅱ),at the same time DHU oxidation occurred.
dihydroxyurea;Fe(Ⅲ);coordination;redox;reaction kinetic
O641.4
A
0253-9950(2011)04-0224-06
2010-04-13;
2011-04-11
晏太红(1979—),男,甘肃临洮人,博士,副研究员,核燃料循环与材料专业
