原位聚合法制备压敏型密胺树脂微胶囊
2010-11-27余红敏王正青
徐 健 余红敏 祁 飞 王正青
(华东理工大学化学与分子工程学院,上海,200237)
原位聚合法制备压敏型密胺树脂微胶囊
徐 健 余红敏 祁 飞 王正青
(华东理工大学化学与分子工程学院,上海,200237)
以三聚氰胺-甲醛树脂作为囊壁,采用原位聚合法对无色染料结晶紫内酯进行包囊,制备了压敏型密胺树脂微胶囊。改变了调酸工艺,提出三步调酸法,有效遏止了分散体系在调酸过程中的蓝变,优化了制备工艺条件:乳化剂质量分数为 50%,乳化在 2000 r/min下分散 20 min,壁材和芯材质量比为 1∶2,固化阶段 pH值为 4.0。结果表明,制备的微胶囊单壳单壁,外表面光泽致密,平均粒径较小,粒径分布集中 (0.5~5.0μm),对芯材包覆完全。
三聚氰胺-甲醛树脂;苯乙烯马来酸酐;原位聚合;压敏型密胺树脂;微胶囊
微胶囊技术是一种隔离和保护囊内染料的保护技术,它采用成膜材料将一些具有反应活性、敏感性或挥发性的液体或固体包封形成微小胶囊[1]。压敏显色微胶囊是无碳复写纸的核心组成部分,常见的制备工艺有复合凝聚法、界面聚合法和原位聚合法[2]。
壁材可选用阿拉伯树胶、聚氨酯、聚酰胺、脲醛树脂和密胺树脂等。当前以广泛的壁材选择、窄化粒径分布、提高微胶囊的性能及降低成本等为热点的研究课题[3]。此外,随着人们环保意识的提高,对于产品的环境友好性也提出了更高的要求。近年来,通过在固化阶段加入甲醛清除剂[4-5]或一种钙盐的方法[6]来降低微胶囊中游离甲醛的含量。
1954年,美国NCR公司首次利用微胶囊技术开发成功了第一代无碳复写纸[7]。微胶囊的制备工艺很难掌握,特别是囊壁的形成、囊壁壁厚的控制、颗粒直径和均匀度等,都直接影响到无碳复写纸生产的成败[8]。我国于 20世纪 80年代后期从国外引进技术生产无碳复写纸。近年来,我国微胶囊技术的发展速度很快,已有不少相关的专利报道,但大多是关于应用方面[9],关于实验条件对微胶囊性能的影响鲜有报道。目前,无碳复写纸的质量仍存在一些问题,主要是显色密度低,尤其是在低温时显色速度慢;耐磨抗压性差,无碳复写纸中页纸胶囊涂层容易起蓝点;耐老化性能差,显色文字图像容易褪色[10]。
为了保护微胶囊,防止其过早破裂,本实验对囊壁进行了改性,改变传统的调酸工艺,以弱酸三步调节体系酸度,控制预聚体在胶囊表面的沉析速率,使胶囊囊壁得到充分固化。本实验以苯乙烯马来酸酐共聚物为乳化剂,三聚氰胺-甲醛树脂为囊壁,采用原位聚合法,制备了包覆无色染料的压敏显色微胶囊,以期探讨密胺树脂微胶囊制备的新技术。
1 实 验
1.1 仪器和药品
仪器 JSF-400搅拌砂磨分散多用机;单相交流感应电动机;S312数显恒速搅拌器;OMEC LS-POP(Ⅲ)激光粒度分析仪;T2-6u尼康倒置生物显微镜;JY3002电子天平。
药品 三聚氰胺-甲醛预聚体,自制;丙烯酸、NaOH均为分析纯;二芳基乙烷、结晶紫内酯(CVL)、苯乙烯马来酸酐乳化剂 (S MA-520)等均为工业级。
1.2 实验方法
1.2.1 芯材溶液的制备
将一定量 CVL加入到二芳基乙烷中,加热搅拌,90℃保温至 CVL全部溶解,液体澄清,冷却备用。
1.2.2 微胶囊的制备
取一定量系统调节剂苯乙烯马来酸酐溶液,加入少量去离子水,在乳化机上高速搅拌,缓慢加入芯材溶液,进行乳化分散,用生物显微镜观察乳化效果,直至油滴粒径小于 5μm,得到O/W稳定乳液。滴加三聚氰胺-甲醛预聚体,以酸液调节溶液 pH值为6.0,转移溶液至三口烧瓶,加热搅拌,调酸 pH值为 5.5,保温 2 h,继续调酸至 pH值为 4.0,于 70℃保温 2 h,停止加热搅拌,陈化 24 h后,出料,调至乳液 pH值为 8。
1.3 测试方法
微胶囊密封性的检测:取少量微胶囊乳液,稀释后涂布于无碳复写纸的 CF纸,烘干后观察,若无显色,则表示微胶囊密封性良好,芯材包覆完全。
微胶囊的囊壁强度测试:取一定量微胶囊粉末、膨化淀粉、聚乙烯醇缩丁醇等,混合均匀,制成无碳复写纸的 CB纸,与 CF纸配合显色,分别测试 6000 N/cm2压强以下的偶然显色和 6000 N/cm2压强以上的书写显色密度。
微胶囊粒径的测量:将微胶囊乳液稀释后超声分散 3 min,用激光粒径分布仪测量平均粒径及粒径分布。
微胶囊表面形貌:取少量微胶囊乳液,稀释后用生物显微镜观察,并拍照。
2 结果与讨论
2.1 乳化速度和乳化时间对分散油滴粒径的影响
微胶囊的平均粒径和粒径分布是微胶囊性能的基本参数。压敏显色微胶囊的粒径一般在 10μm以下,且要分布集中,以便在压力作用下胶囊同时被压破,包覆的芯材瞬时流出与 CF纸发生显色作用,而芯材的乳化效果直接影响到微胶囊的成囊状态。图1为乳化速度和乳化时间对分散油滴平均粒径的影响。
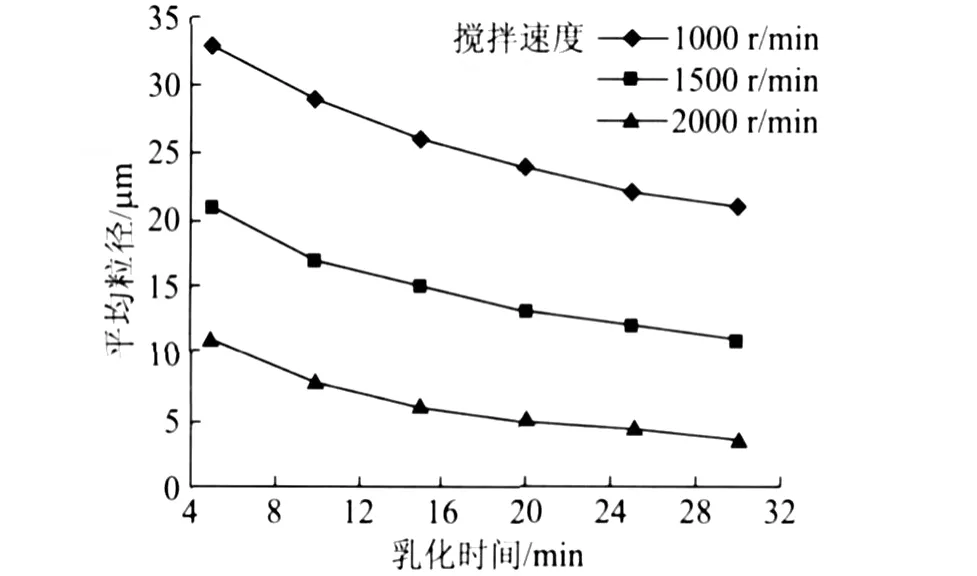
图1 乳化速度和乳化时间对油滴平均粒径的影响
由图1可知,在高速剪切力作用下,起初阶段油滴粒径总体变小,随着时间的延长油滴粒径不再变小而趋于均匀;适当增加搅拌速度可以减少油滴粒径达到平衡需要的时间。在 2000 r/min下乳化 20 min,油滴平均粒径在 3.0μm。图2为该乳化条件下制备的微胶囊粒径分布图。
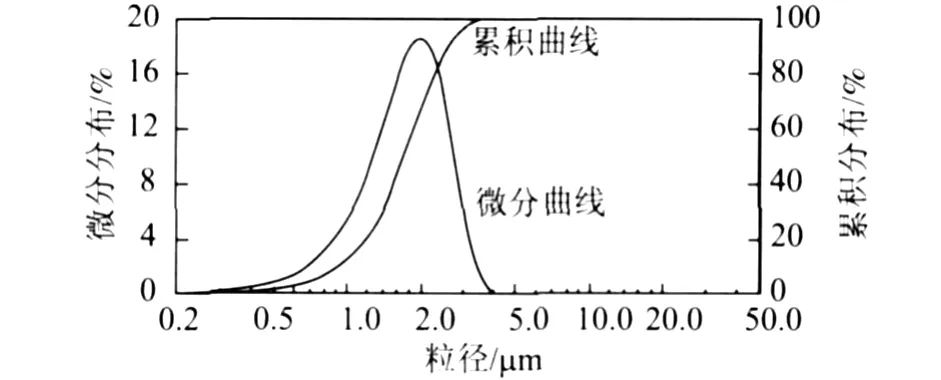
图2 微胶囊的粒径分布
微胶囊的粒径分布均匀,集中在 1.0~4.0μm,平均粒径为 2.6μm,微胶囊的粒径分布情况与乳化后油滴的粒径分布情况相似。因此,只要有适量的乳化剂,能够保持分散油滴在造壁过程中的稳定性,乳液油滴的粒径分布情况直接决定了微胶囊的粒径大小和粒径分布。
2.2 乳化剂用量对微胶囊粒径的影响
分散乳液存在很大的界面,体系的总界面能较高,有明显的凝并倾向。加入 S MA聚电解质溶液,S MA在界面吸附,形成界面膜 (见图3)。界面膜的强度和紧密程度是影响分散油滴稳定性的关键因素。
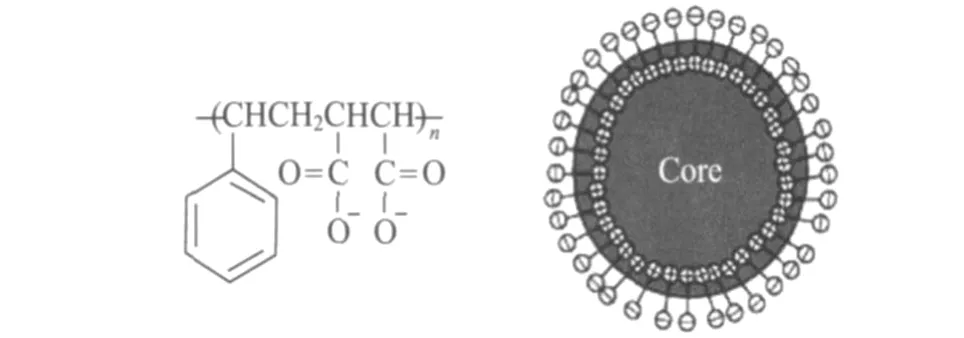
图3 S MA阴离子结构式及在油滴表面的定向排列
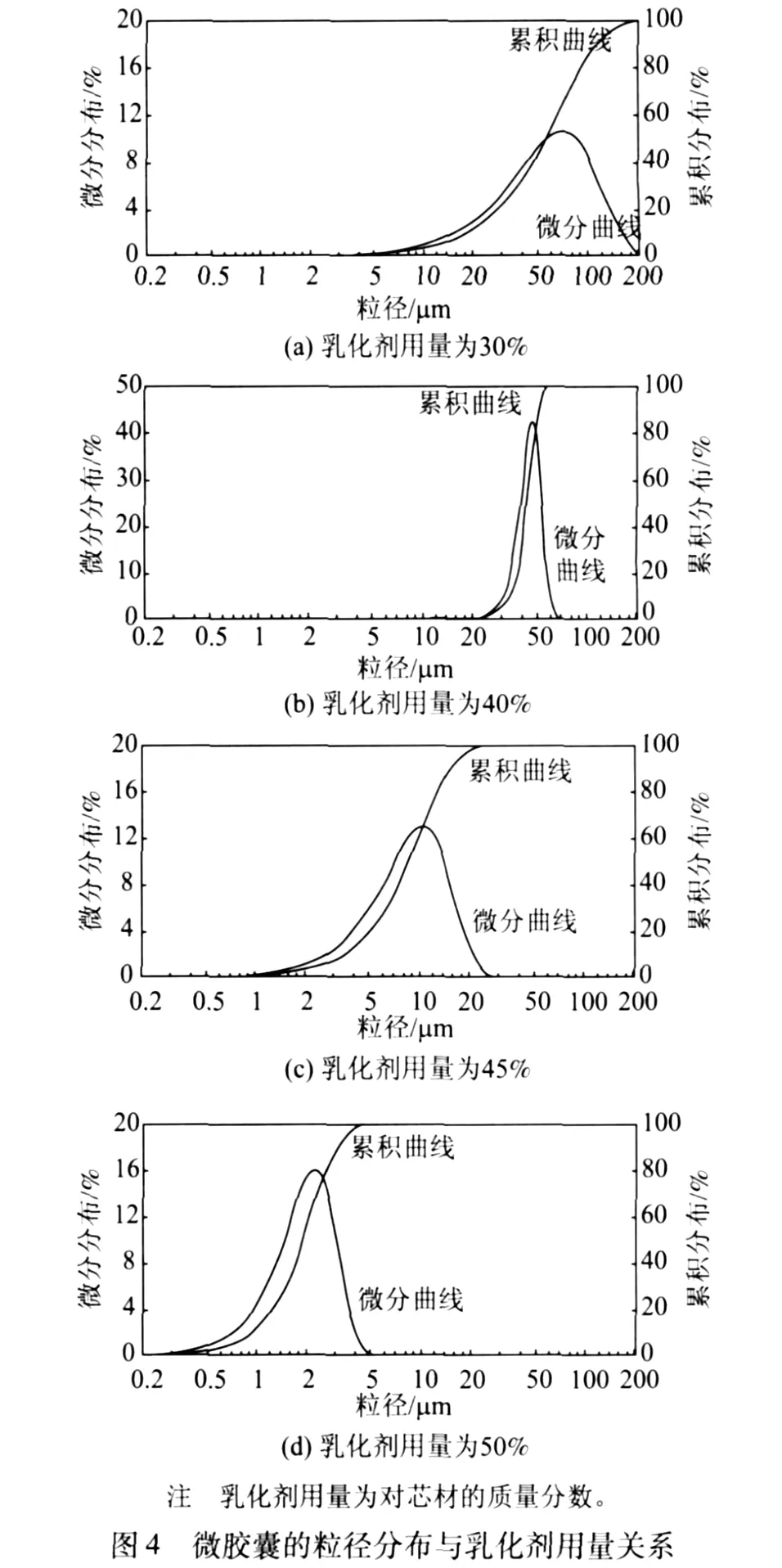
当乳化剂用量过少时,界面膜中分子排列疏松,不能产生足够的立体屏障效应以起到空间稳定的作用,制备的微胶囊粒径过大,且分布不均,微胶囊之间黏结现象严重,涂布于 CF纸上渗色明显;提高乳化剂用量时,微胶囊平均粒径明显变小,分布变窄,微胶囊球形规整度变好,黏连现象逐渐消失;乳化剂用量 (质量分数)为 50%时,微胶囊的粒径分布在 0.5~5.0μm,密封性好,涂布时不易渗色。不同乳化剂用量条件下微胶囊的粒径分布如图4所示。
乳化分散剂除了能保持芯材在连续相中稳定分散外,还能有效地防止因壁材的加入而引起的破乳现象[11]。乳化分散剂的另一个重要功能是能控制或改善壁材预聚体的聚合反应,以获得粒径分布相对集中的微胶囊[12]。
2.3 固化反应 pH值对微胶囊表面形貌的影响
溶液的 pH值直接影响三聚氰胺-甲醛预聚体的缩聚反应。密胺预聚体中具有刚性的羟甲基易在酸性条件下发生缩合,生成稳定的亚甲基键,在加热固化后形成体型网状结构,沉析在液滴表面形成囊壁。不同 pH值对固化反应微胶囊表面形貌的影响如图5所示。
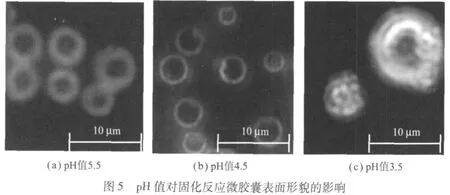
溶液固化反应 pH值在 6.0之上,密胺预聚体缩聚极慢,难以形成微胶囊;溶液 pH值在 5.5时,微胶囊表面疏松,耐压性较差,包覆的芯材容易渗漏;固化反应 pH值在 4.0~4.5时,密胺预聚体缩聚完全,制备的微胶囊强度和密封性较好,表面光泽,涂布不渗色;溶液 pH值为 3.5时,缩聚反应过快,形成的囊壁粗糙,表面吸附大量固体颗粒,微胶囊致密性很差;当调酸用量进一步增加时,体系中富集过多的 H+,极易与芯材中的 CVL接触,导致内酯环的开裂而提前显色。
2.4 壁材与芯材的质量比对微胶囊表面形貌的影响
固定制备工艺条件,改变壁材与芯材的质量比,观察对微胶囊粒径和成囊性能的影响,其表面形貌如图6所示。
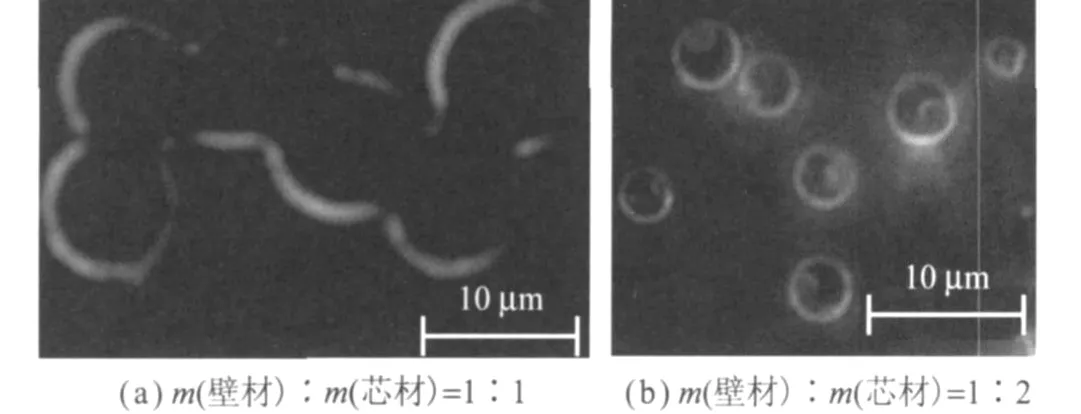
图6 壁材与芯材的质量比对微胶囊表面形貌的影响
壁材与芯材的质量比为 1∶1时,微胶囊的粒径由于壁厚增加而变大,分布在水相中壁材浓度增加,不能保证壁材在分散介质和界面之间有足够的浓度差,致使部分水相中的低聚物交联缩聚后形成未包覆的芯材的树脂颗粒,并吸附在微胶囊表面,造成微胶囊表面粗糙,同时由于体系黏度增加,微胶囊也极易相互黏连;壁材与芯材的质量比在1∶2时,制备的微胶囊密封性和球形规整度较好,粒径分布窄,耐压强度适宜,涂布不渗色;壁材与芯材的质量比进一步增大时,由于壁材减少,形成的囊壁过薄,微胶囊致密性下降,其强度和耐压性变差,在搅拌或涂布过程中容易破裂。
2.5 调酸方式与酸化滴加时间对制备工艺的影响
CVL是一种变色非常灵敏的内酯结构的无色染料,遇酸内酯环断裂,选择性地吸收可见光,呈现相应的颜色。壁材的固化阶段必须在酸性条件下才能进行,实验中既要调节反应体系的 pH值,又不至于使CVL提前变色。因此调酸方式是微胶囊制备过程中的关键环节。
用强酸调节,体系的 H+过于集中,导致体系蓝变絮状,破坏了乳化体系;采用酸性太弱物质(如氯化铵),则耗量过大,费时费力;实验采用质量分数为 4%的丙烯酸调节酸度,酸性适中。实验发现,随着反应时间的延长,体系酸度逐渐降低。
为保证密胺树脂在油滴表面均匀沉积和体系不发生蓝变,成囊时应严格控制 pH值的变化速率。调酸采用滴加方式,并适当延长滴加时间。芯材乳化分散后先调酸至 pH值为 6.0,加热搅拌后再缓慢调酸至pH值为 5.5。保温一段时间后,由于缩聚反应引发后囊芯表面被简单固化,调酸物质加入后不易使囊芯发色,可以调至 pH值为 4.0。因此,调酸方式为3次分批滴加,酸化滴加时间分别为 1 h,0.5 h,2 h。
3 结 论
3.1 以苯乙烯马来酸酐共聚物作为乳化剂,与传统的分散剂相比,如十二烷基苯磺酸钠、吐温系列等,这种阴离子电解质具有更好的在分散体系中稳定分散体、助乳化和保护微胶囊等作用,使微胶囊的粒径大小及分布容易控制。改变了以往的调酸方法,以三步调酸方式,控制酸化滴加时间分别为 1 h,0.5 h,2 h,不易出现因调酸方式不当带来的体系蓝变问题,同时微胶囊囊壁得到充分固化,囊壁厚度和强度可控,易于工业化。
3.2 优化了以密胺树脂为囊壁的 CVL染料微胶囊的制备工艺条件:乳化剂用量 (质量分数)为 50%,乳化条件在 2000 r/min下分散 20 min,壁材与芯材的质量比为 1∶2,固化反应 pH值为 4.0。
3.3 经激光粒径分布仪和生物显微镜分析表明,制备的微胶囊表面光泽,球形规整,无黏连现象,粒径分布均匀 (0.5~5.0μm)。微胶囊的囊壁强度和密封性良好,涂布于无碳复写纸下页纸 (CF纸)上无任何渗色,囊壁压迫后显色明显。
[1] 许时婴,张晓鸣,夏书芹.微胶囊技术[M].北京:化学工业出版社,2006.
[2] Kreysa G,Medin H.Indirect electrosynthesis ofρ-methoxybenzaldehyde[J].Journal ofApplied Electrochemistry,1986,16(5):757.
[3] 陈 影,韩 卿.无碳复写纸的涂料及发展[J].西南造纸,2006,35(2):14.
[4] Gabriele F,Ralf B,Mattias K.Microcapsules of low-formaldehyde melamine/fo rmaldehyde resins:US,6261483[P].2001-07-17.
[5] Dietrich H,Herbert E.Low-viscosity,melamine-formaldehyde resin microcapsule dispersions with reduced formaldehyde content:US,6719931[P].2004-03-13.
[6] 艾泽曼 H,内尔沃 J,比亚斯托驰 H,等.三聚氰胺-甲醛树脂的甲醛降低的微胶囊分散体:中国,101205310[P].2008-06-25.
[7] Green B K.Oil-containing microscopic capsules and method of making them:US,2800458[P].1957-07-23.
[8] 关道生,张世楷.无碳复写纸用微胶囊研制和开发[J].中国造纸,1996,15(6):57.
[9] 甄朝晖,陈中豪,林德森.微胶囊制备技术及其在造纸印刷工业中的应用[J].上海造纸,2006,25(6):18.
[10] John K R.Recording paper incorporating hollow sphericalplastic pigment:US,5932515[P].1999-03-03.
[11] Nzudie D T,Dimonie V L,Sudol E D,et al.Use of styrene-maleic anhydride copolymers(S MA resins)in emulsion copolymerization[J].Appl.Polym.Sci.,1998,70(13):2729.
[12] 甄朝晖,陈中豪.乳化剂对原位聚合密胺甲醛树脂微胶囊成囊性的机理研究[J].中国造纸学报,2006,21(1):47.
Preparation of Pressure SensitiveM elam ine-formaldehyde Resin M icrocapsules by in-situ Polymerization
XU Jian*YU Hong-min Q I Fei WANG Zheng-qing
(Departm ent of Chem istry and M olecular Engineering,East China University of Science and Technology,Shanghai,200237)
The pressure sensitivemelamine-for maldehyde resinmicrocapsuleswere prepared by using in-situ polymerizationmethodwith leuco dye crystal violet lactone as core and the melamine-formaldehyde resin as cyst wall.The three-step method was used to adjust the acidity,and blue-stain of the dispersion system was effectively eliminated in the process.The optimum emulsification conditionswere as follows:dosage of emulsifierwas 50wt%,emulsification was carried out by dispersing for 20 min with 2000 r/min agitation,the suitable proportion of core material and wallmaterialwas 1∶2,the pH of curing phase was 4.0.The results showed that the prepared microcapsules were single core and single wall,outer surface was gloss and compact,average partical size was small,particle size distribution was concentrated at 0.5~5.0μm,the core materialwas fully coated.
melamine-formaldehyderesin;styrene-maleic anhydride;in-situ polymerization;microcapsules
TS727+.3
A
0254-508X(2010)12-0034-04

徐 健先生,在读硕士研究生;主要研究方向:压敏纸显色剂、功能性乳化剂及微胶囊的制备研究。
(*E-mail:030080529@mail.ecust.edu.cn)
2010-08-26(修改稿)
(责任编辑:赵旸宇)
