芳烃污染空气光催化净化材料研究进展与展望
2010-10-24王绪绪付贤智
陈 旬,王绪绪,付贤智
(福州大学光催化研究所,国家环境光催化工程技术研究中心,省部共建光催化国家重点实验室培育基地,福州350002)
芳烃污染空气光催化净化材料研究进展与展望
陈 旬,王绪绪,付贤智
(福州大学光催化研究所,国家环境光催化工程技术研究中心,省部共建光催化国家重点实验室培育基地,福州350002)
芳烃化合物是一类难降解的有毒挥发性有机物,排放到大气对生态环境有极大的危害。光催化技术作为一种高级氧化技术是未来环境净化技术的发展方向之一。然而,经典的TiO2材料对芳烃的光催化氧化,不仅活性低而且极易失活,这在很大程度上制约了光催化技术在空气净化领域的实际应用。在综述国内外研究进展的基础上,简要总结了最近几年我们在芳烃污染空气的光催化净化材料开发和性能研究方面的一些进展。研究结果显示,有众多宽带隙半导体材料对芳烃表现出比TiO2更加优异的光催化活性和活性稳定性;有些材料甚至在可见光诱导下可表现出非常高的光催化活性。此外,在反应气氛中添加少量氢气可以大幅度提高贵金属修饰TiO2对苯的光催化降解效率。这些进展充分显示出光催化技术在工业三苯废气处理和空气净化等方面的广阔应用前景。
进展;材料;光催化;空气净化;芳烃
前 言
环境污染与能源危机是威胁人类生存和社会经济可持续发展的两大全球性问题。在众多环境污染源中,化学污染最普遍、最主要以及对人类赖以生存的环境,包括水资源、土壤和大气环境的影响最大。其中,苯系化合物,特别是苯、甲苯和二甲苯是一类高挥发性、结构稳定、污染渠道广泛、毒性大而难降解的空气污染物。《Science》最近发表的一篇研究报告[1-2]以我国制鞋业为例详细论述了苯污染对人和动物危害的严重性,指出即使空气中含有1μL/L浓度的苯也会给人造成严重危害。因此,关于空气中微量苯及其衍生物的消除,引起了人们高度重视。对于包括苯系污染物在内的有机废气污染,传统的处理技术主要有催化燃烧法、活性炭吸附法和液体吸收法等。催化燃烧法是在高温下通过催化剂使有害气体催化燃烧成CO2和H2O而消除,但催化剂寿命短,投资费用高,能耗大。吸附法和吸收法,由于吸附剂或吸收剂易达到饱且需要经常再生,有二次污染,不但运行费用高而且操作和管理也比较复杂。对于微量芳烃污染,这些方法都难于实际应用,且只能用于室外集中处理,很难用于室内空气的净化。研制开发高效、低成本、持久耐用的新技术和系统,有效治理空气中微量芳烃污染,对于各种有芳烃污染的行业以及普遍存在的机动车量排放造成的大气污染净化有重大实际意义。
自从上世纪70年代发现TiO2的光催化现象以来,以光催化分解水制氢为目标的能源光催化和以空气和水净化为目标的环境光催化成为能源、催化、材料和环境等科学领域研究的热点。环境光催化依据半导体材料在光激发下能活化分子氧和水分子产生强氧化能力的自由基,进而可以把各种有机分子彻底矿化的原理,被认为是一种高级氧化技术和高效氧化消除各种微量有机污染物一条绿色途径。近二十多年的大量基础和应用研究已经展现出光催化作为新型环保技术的广阔应用前景,目前已经在室内空气净化和染料废水处理方面获得一些应用。光催化技术与传统技术相比具有无可比拟的优点:高效、彻底以及有可能利用太阳光而低能耗、绿色。因此采用光催化技术来治理空气中微量芳烃污染无疑是未来空气净化技术发展方向和理想选择。
早在上世纪,就已经有大量关于T iO2在微量芳烃光催化消除的研究报道[3-6],表明通过T iO2的光催化作用能够有效地将水中或空气中芳烃分子氧化成CO2和水,近二十多年来,相关研究报道不断。然而,大量研究毫无例外地发现,与其它的挥发性有机污染物(VOCs)如三氯乙烯、甲醛等等相比,芳烃VOCs在T iO2上的光催化反应速率很低,且在反应过程中催化剂失活严重[7-8]。有人详细研究了空气中甲苯在TiO2上连续流动光催化过程,表明反应开始甲苯的消除转化率达到75%,但反应3 h后转化率迅速降低到20%,同时催化剂颜色变黑[9]。也有人考察了循环反应模式下苯在T iO2上的光催化氧化,发现催化剂同样失活严重[10]。我们在连续流动催化反应装置上考察了P25 TiO2对苯的光催化行为,发现在我们的实验条件下苯的光催化转化率不到9%,且6 h后催化剂活性完全丧失。为解决T iO2光催化芳烃氧化活性低和易失活的问题,国内外研究者也开展了不少研究,如发现用贵金属修饰TiO2[11]和给反应气氛中添加水蒸气[12]可一定程度地改善光催化过程效率和催化剂活性稳定性。然而,由于TiO2催化剂对芳烃的光催化氧化转化率很低,即使有较稳定的活性或者采取措施能延缓催化剂失活,也难于从根本上解决问题和满足实际应用的要求。
很显然,欲实现光催化技术在含芳烃VOCs净化上的实际应用,必须研制新型高效光催化材料。要求催化剂不但有高的光催化活性,还应有高的活性稳定性或抗失活性,同时希望能实现在可见光下苯的降解。最近几年,我们瞄准这一难点问题开展了大量研究,探索研究了一系列新光催化材料或体系。本文将重点介绍在这方面取得的一些进展。
新型紫外光光催化材料
. β
Ga2O3作为光解水制氢催化材料曾经有过报道[13-14]。我们以Ga(NO3)3、乙醇和浓氨水为原料并采用共沉淀法、真空干燥和600℃焙烧等步骤,制备了一种具有介孔结构的β-Ga2O3。表明这样制备的β-Ga2O3具有4.7 eV的带隙,对紫外光有强吸收,且价带电位比T iO2正(图1),在紫外光(254 nm)下它对含在空气中的芳烃(苯、甲苯)表现出比TiO2优异得多的光催化氧化性能和活性稳定性,苯的降解转化率接近达到60%,且转化的苯接近完全矿化(图2)[15]。

图1 β-Ga2O3和TiO2(锐钛矿)的紫外漫反射吸收光谱(a)和能带位置(b)Fig.1 UV-Vis diffuse reflectance absorption spectra(a)and the energy levels(b)of conduction band edge and the valence band edge with respect to absolute vacuum scale of TiO2and Ga2O3

图2 Ga2O3和TiO2对苯(a)和甲苯(b)的光催化降解活性Fig.2 Photocatalytic performances of Ga2O3and TiO2for decomposing benzene(a)and toluene(b)in air(initial concentrations of benzene and toluene in the stream are 450 and 350μL/L,respectively)
进一步采用沉淀法制备了α-,β-和γ-相Ga2O3[16-17]。比较发现,在254 nm的紫外光诱导下,这3种晶相Ga2O3对苯系污染物的消除能力有明显的差别,按照β-Ga2O3>α-Ga2O3>γ-Ga2O3顺序依次降低,但性能均优于TiO2。通过晶相结构、织构和光生载流子产生量的分析比较推断,材料的结晶度和几何结构是影响Ga2O3光催化性能的主要原因,β-Ga2O3因具有高结晶度和畸变的几何结构,而表现出最高的光催化活性和活性稳定性。
. 纳米晶
In(OH)3也是一种禁带宽度约为5.15 eV的半导体材料,但作为光催化剂却很少被研究。我们采用超声解胶技术制备了小粒径、高比表面的In(OH)3纳米晶,以气相中的苯和甲苯为目标污染物,考察了其光催化性能(图3)。发现在254 nm的紫外光照射下,与P25 TiO2相比,In(OH)3对苯和甲苯均表现出了很高的光催化活性和活性稳定性。对浓度为1 800μL/L含苯空气,苯的稳态光催化转化率和矿化率分别可达到33%和56%,而且基本保持活性稳定。发现引入水蒸气可以进一步改善In(OH)3对苯和甲苯的光催化活性持久性。认为In(OH)3之所以对苯的降解具有高活性和高活性稳定性,可能与In(OH)3具有较高的空穴氧化能力和表面不易结碳有关。

图3 In2O3和P25 TiO2对苯的光催化降解活性比较Fig.3 Activities of In2O3and P25 TiO2for the benzenephotocatalytic degradation
. 纳米晶
InOOH纳米晶作为光催化剂是我们首次报道。它采用溶剂热法,以In(NO3)3·4.5H2O为原料在反应釜内的H2O/乙二胺中180℃制备。所合成的InOOH样品呈现良好分散的不规则球状纳米颗粒,平均尺寸大约20 nm,BET比表面积为55 m2/g。从紫外漫反射光谱(图4)估计其带隙为3.7 eV。InOOH在300 nm的紫外光下对苯光催化稳态转化率为7.5%,矿化率为30%,而且有高活性稳定性,性能明显优于P25 TiO2(图5)。?
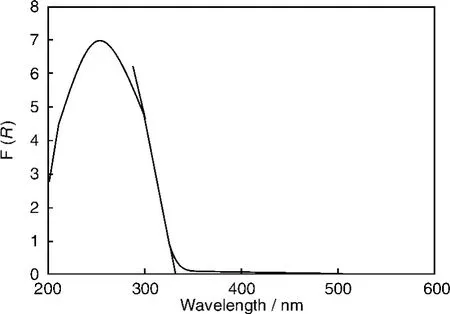
图4 InOOH紫外漫反射吸收光谱Fig.4 UV-Vis diffuse reflectance absorption spectrum of InOOH
ZnSn(O H)6(ZHS)是一种钙钛矿结构的氢氧化物,结构类似于In(OH)3。这个材料可以用各种方法合成,但尚未见到有关作为光催化材料的研究报道。我们以ZnCl2,SnCl4,n-C4H9NH2为原料,在乙二醇水溶液中通过水热法制备出了具有立方晶体形貌的ZnSn(OH)6。研究表明,这样制备的样品,呈立方晶体形貌,结晶良好(图6);光吸收截止波长为290 nm,对应于4.3 eV的禁带宽度(图7);有48 m2/g比表面积。ZnSn(OH)6对苯表现出比P25 T iO2高的光催化降解效率和稳定性,苯的转化率为大约17%,矿化率约为5%(图8)。实验证明,羟基自由基和超氧自由基均为这个反应中的活性中间体。其具有良好活性的原因可能与样品有大量羟基有关。

图5 InOOH和TiO2对苯的光催化降解活性比较Fig.5 Comparison of photocatalytic activities of InOOH and TiO2for benzene degradation

图6 ZnSn(OH)6的SEM照片Fig.6 SEMmicrograph of ZnSn(OH)6
. 纳米棒
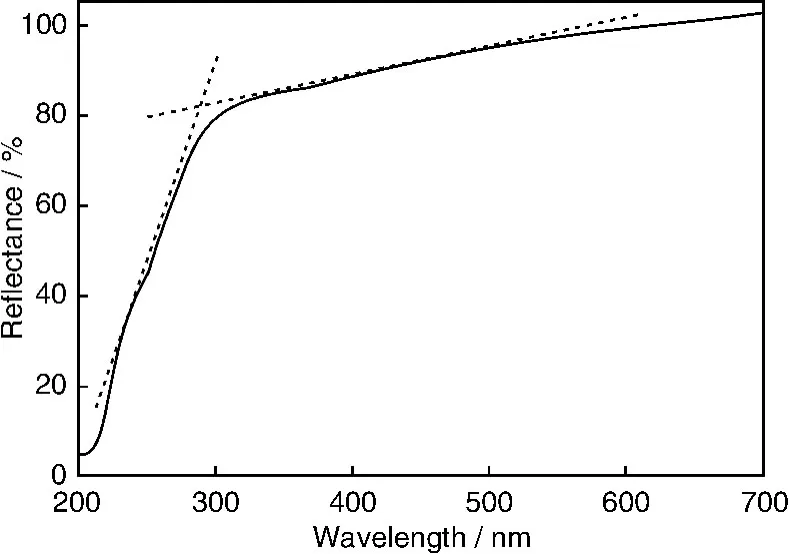
图7 ZnSn(OH)6的紫外漫反射吸收光谱Fig.7 UV-Vis diffuse reflectance absoiption spectrum of ZnSn(OH)6

图8 ZnSn(OH)6(a)和P25 TiO2(b)对苯的光催化降解活性比较Fig.8 Comparison of benzene concentration and the amount of the CO2produced for ZnSn(OH)6(a)and P25 TiO2(b)as a function of irradiation time underUV illumination(λ=254 nm),fow rate of the reactantmixture 40 mlmin-1

图9 Zn2GeO4纳米棒的SEM照片Fig.9 SEMmicrograph of Zn2GeO4nanorods
二元金属酸盐锗酸锌(Zn2GeO4)作为光催化分解水产氢催化材料已有报道[21]。我们[22]采用表面活性剂助水热法合成出了一种纳米棒状的Zn2GeO4(图9)。该样品经表征具有4.7 eV的禁带宽度,BET表面积36 m2/g,光催化性能大大地优于体相Zn2GeO4,TiO2(P25),Pt/TiO2。在连续流动反应模式下,Zn2GeO4纳米棒对苯的稳态光催化转化率达21%,矿化率达75%,在120 h的连续反应后其活性依然是稳定的(图10)。对其它挥发性芳香烃污染物(甲苯、乙苯等),其同样表现出优异的光催化性能。电子自旋捕获EPR表征表明,在紫外光照射下Zn2GeO4纳米棒在水中能产生大量羟基自由基,推断降解苯的过程可能按照羟基自由基诱导机理进行。

图10 Zn2GeO4纳米棒和TiO2对苯的光催化降解活性比较:(a)苯的转化率,(b)CO2产量Fig.10 Photocatalytic conversion ratio of benzene(a)and concentration of produced CO2(b)in the stream for the Zn2GeO4nanorods versus the reaction time
. 纳米晶
Sr2Sb2O7是一种P型宽带隙半导体材料,同样已经被作为水分解制氢光催化材料研究过[23]。我们以Sb2O5和Sr(CH3COO)2·0.5H2O作为Sb和Sr源,通过常规水热法合成出了平均晶粒直径约6 nm,禁带宽度约4.2 eV,BET比表面积25m2·g-1的Sr2Sb2O7纳米晶体[24]。此外,还采用文献中报道的常规固态反应法用SrCO3和Sb2O3作原料合成出了大晶粒的Sr2Sb2O7(SSR)样品。比较研究发现,这两个样品在光吸收和光催化性能上有明显的差别。前者在UV-vis漫反射光谱上观察到显著的纳米尺寸效应—光吸收波长蓝移(图11)。在连续流动反应模式下在254 nm的光激发下,苯在P25 TiO2上的光催化降解转化率在反应初期仅为10.5%,且随着反应进行很快降低至4.2%,同时只有少量CO2生成(图12)。而在同样条件上,用水热法制备的Sr2Sb2O7纳米晶体样品具有24%的稳态转化率以及50%的矿化率,且在40 h的反应中活性保持稳定。至于用固态化学法制备的Sr2Sb2O7(SSR)样品,活性虽然稳定,但苯的稳态苯转化率只有4%,CO2的产量也比较低。

图11 两个不同方法所合成Sr2Sb2O7样品的紫外漫反射吸收光谱Fig.11 UV-Vis diffuse reflectance absorption spectra of Sr2Sb2O7(180℃,48 h,[OH-]=2 mol·L-1)and Sr2Sb2O7(SSR)

图12 Sr2Sb2O7,Sr2Sb2O7(SSR)和TiO2对苯的光催化降解活性Fig.12 Photocatalytic activity of Sr2Sb2O7,Sr2Sb2O7(SSR)and TiO2for the degradation of benzene
. 锗酸镉纳米晶[]
Cd2Ge2O6晶体被表明也是一种对苯光催化降解具有良好性能的光催化材料,其以CdAc2和GeO2为原料通过水热法合成。Cd2Ge2O6具有类似于Zn2Ge2O6的棒状形貌,光吸收随着水热温度的增加而发生红移,带隙随之变窄(图13)。100℃温度下所合成样品的光吸收边在大约260 nm,对应于4.8 eV的带隙能,而140和180℃得到的样品分别具有322和325 nm的吸收边,对应于约3.9 eV的带隙。在254 nm紫外光照射下,这两种样品对苯都表现优于TiO2的光催化活性,但100℃所制Zn2Ge2O6的活性最好(图14),140℃制备的样品的活性次之,180℃制备的样品的活性最差。100℃制备Zn2Ge2O6材料对苯的降解转化率高达~60%,而且活性在短时间内可达到稳态。其高活性的原因可能与独特的电子结构和大的比表面积有关。
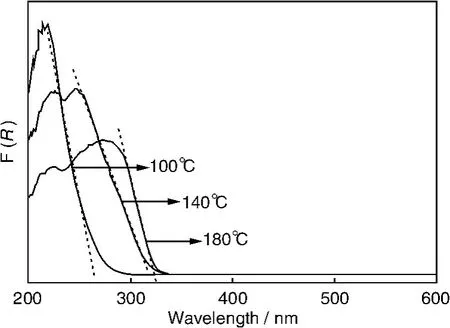
图13 不同水热温度所制Cd2Ge2O6的紫外漫反射吸收光谱Fig.13 UV-Vis diffuse reflectance absorption spectra of preparedsamples at different temperature
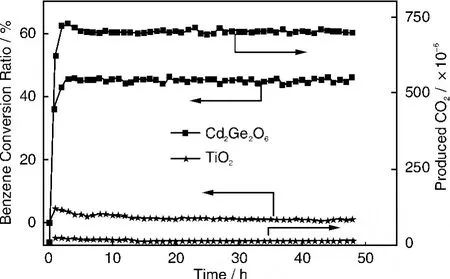
图14 Cd2Ge2O4对苯的光催化降解活性Fig.14 Photocatalytic activity of Cd2Ge2O4for benzene degradation in continuous flow mode
. 镓酸锌多孔纳米晶[]
ZnGa2O4也是一种宽带隙半导体,光吸收截止波长在大约320 nm。我们以Ga(NO3)3和Zn(NO3)3为原料,采用水热法制备出了一种多孔结构的ZnGa2O4纳米晶体。表1列出了在不同水热温度所制ZnGa2O4的织构参数。表明样品的比表面积随水热晶化温度的升高急剧下降,孔径和晶粒尺寸增大,孔隙率下降。80℃水热制备的样品织构最好,具有200 m2/g的比表面积和大约1.7%孔隙率。图15是不同温度制备样品以及TiO2对苯光催化降解性能的比较。可以看出,ZnGa2O4样品的活性均优于P25 TiO2,其中80℃水热制备样品的活性最高,这可能与其大比表面积、多孔性有关。此外,反应80 h得到的样品对于苯的降解的转化率基本保持>41%的转化率和>500μL/L的CO2生成量。此外研究表明,80℃水热制备ZnGa2O4样品对甲苯和乙苯同样具有高的光催化降解活性和活性稳定性,甲苯的稳态转化率大于41%,乙苯的稳态转化率高于30%。很显然,这是一种非常优良的苯光催化降解光催化材料。

表1 不同水热温度所制ZnGa2O4纳米晶体的组织参数Table 1 Texture pa ram e te rs of ZnGa2O4

图15 不同水热温度所制ZnGa2O4对苯的光催化降解性能Fig.15 Photocatalytic conversion of benzene(a)and the concentration of produced CO2(b)over different ZnGa2O4catalysts under 254 nm illumination
新型可见光光催化材料
前面列举的这些光催化材料在紫外光下对苯都有很好的光催化降解性能,而实现在可见光下对苯的高效降解显然具有更重要的实际意义。可见光响应的光催化材料一直是近年来国际光催化研究的重点。然而,对于芳烃VOCs的可见光降解,相关光催化材料的研究报道并不是很多,主要原因是TiO2基催化剂对苯都没有很好的降解性能。近年来我们一直致力于这方面的研究,取得了一些进展,发现由矾酸盐与T iO2组成的双组分复合材料对降解苯是一种非常优良的可见光光催化材料。
. 敏化的材料
InVO3是一种具有带隙2.1 eV的半导体,早先作为光解水催化剂被研究[27]。我们采用溶胶-凝胶法将0.5%InVO4与TiO2进行复合制得一种InVO3敏化的T iO2材料[28]。这种材料具有介孔结构,InVO4与TiO2以异质结结构复合(图16)。从UV-Vis漫反射光谱(图17)观察到,复合样品在可见光区有显著的吸收,光吸收截止波长扩展到540 nm。研究表明,其在450-900 nm的可见光照射和气体流速为50 cm3/min的反应条件下,对于浓度为200μL/L的苯、甲苯、乙苯、环己烷和丙酮分别可达到50%,57%,64%,62%,31%的稳态光催化氧化转化率,12 h反应未见明显失活。(图18)。该催化材料的光催化活性可以很好地基于In-VO3可见光敏化TiO2机理进行解释,InVO4和TiO2具有相互匹配的能带结构,光生电子能有效地从InVO4导带向TiO2导带转移,实现可见光敏化。

图16 InVO4/TiO2的TEM(a)和HRTEM(b)照片Fig.16 TEM(a)and HRTEM(b)micrographs of InVO4/TiO2sample

图17 几种光催化样品的紫外漫反射吸收光谱Fig.17 UV-Vis diffuse reflectance absorption spectra of several photocatalytic samples

图18 可见光下InVO4/TiO2(a),TiO2-xNx(b),InVO4(c),TiO2(d),InVO4/SiO2(e)光催化降解苯活性(A)和InVO4/TiO2降解苯的活性稳定性(B)Fig.18 Photodegradation(A)of benzene(215μL·L-1)undervisible light irradiation on InVO4/TiO2(a)TiO2-xNx(b),InVO4(c),TiO2(d),InVO4/SiO2(e)and activity stability(B)of InVO4/TiO2for benzene degradation under visible illumination
. 敏化的材料[]
从2.1节的实验结果可以看出,窄带隙半导体与TiO2进行异质结复合是拓宽TiO2可见光吸收的有效途径。我们进一步采用简单的水热法制备了稳定的窄带隙(~2.1 eV)多金属氧化物钒酸镧(LaVO4),并采用简单的溶胶-凝胶耦合方法制备了异质结构纳米复合材料LaVO4/TiO2。实验结果表明,LaVO4/TiO2也是具有异质结的纳米材料,能有效吸收可见光,且具有多孔结构和较大的比表面积(80 m2/g)。这种LaVO4/TiO2在可见光照射下对苯的转化率为58%,同时生成约260μL/L的CO2,相应的矿化率约为32%(图19)。经过5轮总共50 h光催化反应,催化剂基本不失活,表现出高效稳定的可见光光催化活性(图20)。另外,在紫外光和模拟太阳光照射下,LaVO4/T iO2也表现出优于纯T iO2的光催化降解苯的活性。
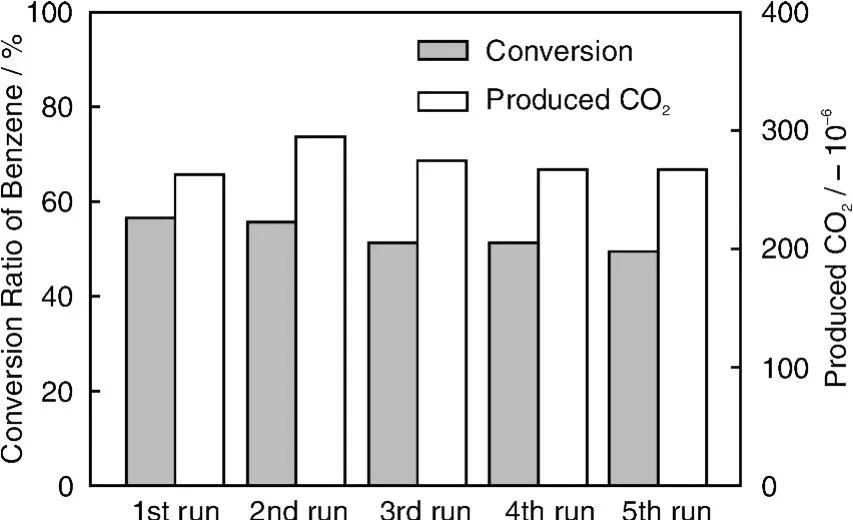
图20 可见光下LaVO4/TiO2降解苯的活性稳定性Fig.20 Activity stability ofLaVO4/TiO2for benzene degradation under visible light irradiation
-耦合高效率光催化系统
. -耦合紫外光光催化体系[]
气相光催化氧化反应一般是以氧气或空气作为载气和氧化剂,然而我们发现,在富氧的反应气氛中添加少量的还原性气体H2,可极大地提高Pt/TiO2催化剂的光催化活性和稳定性。与未加氢气时相比,Pt/TiO2催化剂对苯的光催化氧化分解活性可提高近两个数量级,苯几乎被完全矿化为CO2和H2O,并在长时间的反应过程中催化剂没有发生失活(图21)。H2的最佳添加量在H2/O2约0.2~0.3。对于其它有机污染物,这个体系也同样有效,活性顺序为环己烷<丙酮<苯<二甲苯<乙苯。EPS自由基捕获研究结果表明,Pt和TiO2在光催化过程中共同起作用,H2和O2之间存在耦合效应,H2的加入能有效地抑制稳定的中间产物在催化剂表面形成、表面羟基游离基的数量增加和光生电子-空穴对分离效率的提高,作用机理见图22。

图21 反应气氛对Pt/TiO2上苯光催化降解转化率的影响Fig.21 Effect of different reaction atmospheres on the conversion ratio of benzene(845μL·L-1)on a Pt/TiO2photocatalyst.(H2∶O2=0.02∶1)(a)and effect of the H2∶/O2ratio on the photocatalytic perfor mance of Pt/TiO2toward benzene decomposition(b)
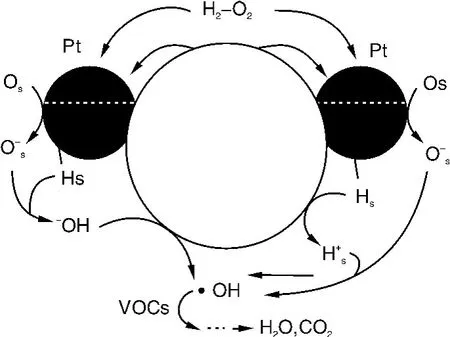
图22 反应机理图示Fig.22 Possible mechanism for the photochemical generation ofOHradicals on a Pt/TiO2catalyst in the coexistence of H2and O2
. -耦合的可见光光催化体系[]
对于H2-O2耦合光催化体系,当用TiO2-xNx代替TiO2作为主催化剂时,可以在可见光下发生光催化。Pt/TiO2-xNx通过改进溶胶-凝胶法和浸渍法制备。图23为TiO2、Pt/TiO2和Pt/TiO2-xNx3个样品的光吸收光谱比较。可以看出,Pt/T iO2-xNx在可见光区有非常显著的吸收,并证明Pt在样品中是高分散的。在可见光照射下连续流动反应装置上的活性评价实验表明(图24),在纯H2气氛下,苯的转化率为零;在纯氧气氛下,苯的转化率只有1%;而在H2-O2混合气氛下,苯的转化率显著升高。当用H2/O2摩尔比为0.02混合气时,苯的转化率可达到18%,矿化率可达到80%,且催化剂活性长时间保持稳定。对其它VOCs,该体系同样有效,但转化率按照环己烷<苯<甲苯<乙苯<丙酮<乙烯顺序增加。其反应机理类似于图22所示。

图23 TiO2、TiO2-xNx和Pt/TiO2-xNx的漫反射吸收光谱Fig.23 Diffuse reflection absorption spectra of TiO2,TiO2-xNx,and Pt/TiO2-xNx

图24 Pt/TiO2-xNx在H2-O2气氛下对苯的可见光光催化降解性能Fig.24 Effects of different reaction atmospheres on the photodegradation of benzene for Pt/TiO2-xNxirradiated by visible light(H2/O2=0.02)
结论与展望
芳烃类VOCs是很难降解的化合物,公认TiO2对其表现出很低的活性和极差的活性稳定性,因而难于实际应用。但是文中所述研究结果显示,同样在紫外光诱导下,有很多材料都表现出比T iO2优异得多的光催化性能。在我们所报道的8种紫外光光催化材料中,β Ga2O3活性最高,对苯的降解有60%的转化率和接近100%的矿化率;其次是Cd2Ge2O6和ZnGa2O4纳米晶体,对苯都有接近50%~60%的降解率和高的矿化率,活性也都非常稳定。此外,In(OH)3,ZnSn(OH)6,Zn2GeO4纳米棒和InOOH也都有高于20%的苯降解转化率和良好的稳定性。这说明,光催化技术的确是一种普适性的高级氧化技术,问题的关键在于针对反应物的特性获得有效的光催化材料。
对两种矾酸盐敏化的T iO2材料——InVO3/T iO2和LaVO3/TiO2的研究发现,它们在可见光下对苯表现出很高的光催化降解活性,比起TiO2在可见光下几乎没有活性是一个重要的进展。或许通过进一步的催化剂和反应条件优化研究,这样的催化剂能满足真正在太阳光诱导下实现芳烃VOCs光催化净化的实际需要。
苯在Pt/TiO2材料上H2-O2耦合高效降解是一个新现象。这样的体系依赖于催化材料在紫外光或在可见光下均表现出很高的光催化效率和活性稳定性,这不但具有重要的实际应用价值,而且对认识光催化过程的微观机理具有重要的理论意义,值得进一步深入研究。
尽管这些催化材料或体系对TiO2难于光催化降解的苯,表现如此优异的光催化活性及活性持久性的原因,我们还不能从理论和机理上给予非常清楚的阐明,但这些结果突破了芳烃分子难于用光催化方法有效降解的难点,为芳烃VOCs污染空气的净化研究注入了新的活力,展现出很大的研究开发空间。
未来的进一步研究仍然非常必要。在基础研究方面,应该结合苯分子的化学结构和电子结构、催化材料的组成、表面结构和电子结构,认识基质-催化材料-反应效率之间的本质联系,阐明这些光催化过程的作用机理,深刻认识控制芳烃VOCs光催化降解的关键因素,如此才能开发效能更好的材料。在应用研究方面,需要进一步优化催化剂组成和结构,优化反应条件,进行工程化研究,为芳烃污染空气净化的实际应用提供基本参数。
我们相信,不久的将来光催化技术一定能在空气污染净化领域获得更加广泛的实际应用。
[1]Stokstad E.Factory Study Shows Low Levels of Benzene Reduce Blood Cell Counts[J].Science,2004,306:1 665-1 668.
[2]Lan Q,Zhang L,Smith M,et al.Hematotoxicity in Workers Exposed to Low Levels of Benzene[J].Science,2004,306:1 774-1 776.
[3]Carey J H,Lawrence J,Tosine H M.Photodechlorination of PCB's in the Presence of Titanium Dioxide in Aqueous Suspensions[J].Bull Environ Contam Toxicol,1976,16:697-701.
[4]Frank S N,Bard A J.Heterogeneous Photocatalytic Oxidation of Cyanide and Sulfite in Aqueous Solutions at Semiconductionor Powders[J].J Phys Chem,1977,81:1 484-1 490.
[5]Norman N,Lichtin,Mahmoud Sadeghi.Oxidative Photocatalytic Degradation ofBenzene Vapor over TiO2[J].J Photochem Photobiol A,1998,113(1):81-88.
[6]Fu X,ZeltnerW A,Anderson MA.The Gas-Phase Photocatalytic Mineralization of Benzene on Porous Titania-Based Catalysts[J].Appl CatalB:Environ,1995(6):209-224.
[7]Lichtin N N,Sadeghi MJ.Oxidative Photocatalytic Degradation of Benzene Vapor over TiO2[J].J Photochem Photobio A Chem,1998,113(1):81-88.
[8]Einaga H,Futamura S,Ibusuki T.Photocatalytic Decomposition of Benzene over TiO2in a Humidified Airstream[J].Phys Chem Chem Phys,1999(1),4 903-4 908.
[9]SauerML,Hale MA,Ollis D F.Heterogeneous Photocatalytic Oxidation of Dilute Touene-Chlirocarbon Mixtures in Air[J].J Photochem Photobiol A,1995,88(1):169-178.
[10]Sitkiewitz S,Heller A.Photocatalytic Oxidation of Benzene and Stearic Acid on Sol-GelDerived TiO2Thin FilmsAttached to Glass[J].New J Chem,1996,20:233-242.
[11]Hisahiro E.Complete Oxidation ofBenzene in Gas Phase by Plati-Nized titania Photocatalysts[J].Environ Sci Technol,2001,35:1 880-1 884.
[12]Einaga H,Futamura S,Ibusuki T.Heterogeneous Photocatalytic Oxidation of Benzene,Toluene,Cyclohexene and Cyclohexane in Humidified Air:Comparison of Decomposition Behavior on Photoirradiated TiO2Catalyst[J].Appl Catal B Environ,2002,38:215-225.
[13]Kudo A,Mikami I.Photocatalytic Activities and Photophysical Properties of Ga2-xInxO3Solid Solution[J].J Chem Soc Faraday Trans,1998,94:2 929-2 932.
[14]Yanagida T,Sakata Y, Imamura H.Photocatalytic Decomposition of H2O into H2and O2over Ga2O3Loaded with NiO[J].Chem Lett,2004,33:726-727.
[15]Hou YD,Wang X C,Wu L,et al.Efficient Decomposition of Benzene over aβ-Ga2O3Photocatalyst under Ambient Conditions[J].Environ Sci Techno,2006,40:5 799-5 803.
[16]Hou YD,Wu L,Wang X C,et al.Photocatalytic Performance ofα-,β-,andγ-Ga2O3for the Destruction ofVolatile Aromatic Pollutants in Air[J].J Catal,2007,250:12-18.
[17]Che Yousan(陈友三),WangXuxu(王绪绪),LiDanzhen(李旦振),et al.高活性低失活In(OH)3纳米晶光催化剂的制备和表征[J].Chem J Chinese Uni(高等学校化学学报),2007,28(2):355-357.
[18]Yan T J,Long J L,Chen Y S,et al.Indium Hydroxide:A Highly Active and Low Deactivated Catalyst for Photoinduced Oxidation of Benzene[J].C R Chim ie,2008,11:101-106.
[19]Li Z,Xie Z,Zhang Y,et al.Wide Band Gap p-Block Metal Oxyhydroxide InOOH:A New Durable Photocatalyst for Benzene Degradation[J].J Phys Chem C,2007,111(49):18 348-18 352.
[20]Fu X L,Wang X X,Ding Z X,et al.Hydroxide ZnSn(OH)6:A Promising New Photocatalyst for Benzene Degradation[J].Appl Catal B Environ,2009,91:67-72.
[21]Sato J,Kobayashi H,Ikarashi K,et al.Photocatalytic Activity for Water Decomposition of RuO2-Dispersed Zn2GeO4with D10 Configuration[J].J Phys Chem B,2004,108:4 369-4 375.
[22]Huang J H,Wang X C,Hou Y D,et al.Degradation of Benzene over a Zinc Germanate Photocatalyst under Ambient Conditions[J].Environ Sci Tech,2008,42:7 387-7 391.
[23]Sato J,Saito N,Nishiyama H,et al.Photocatalytic Watern Decomposition by RuO2-Loaded Antimonates,M2Sb2O7(M=Ca,Sr),CaSb2O6and NaSbO3,with D10 Configuration[J].J Photochem Photobiol A,2002,148(1/3):85-89.
[24]Xue H,Li Z H,Wu L,et al.Nanocrystalline Ternary W ide Band Gap p-Block Metal Semiconductor Sr2Sb2O7:Hydrothermal Syntheses and Photocatalytic Benzene Degradation[J].J Phys Chem C,2008,112:5 850-5 855.
[25]Huang J H,Ding K N,Wang X C,et al.Nanostructuring Cadmium Ger manate Catalysts for Photocatalytic Oxidation of Benzene at Ambient Conditions[J].Langmuir,2009,25(14):8 313-8 319
[26]Zhang X N,Huang J H,Ding K N,et al.Photocatalytic Decomposition of Benzene by Porous Nanocrystalline ZnGa2O4with a High Surface Area[J].Environ Sci Technol,2009,43(15):5 947-5 951.
[27]Ye J,Zou Z,Arakawa H,et al.Correlation of Crystal and Electronic Structureswith Photophysical Propertiesof Water Splitting Photocatalysts InMO4(M=V5+,Nb5+,Ta5+)[J].J Photochem Photobiol A Chem,2002,148(1/3):79-83.
[28]Xiao G C,Wang X C,Li D Z,et al.InVO4-Sensitized TiO2Photocatalysts for EfficientAir Purification withVisible Light[J].J Photochem Photbiol A Chem,2008,193:213-221.
[29]Huang H J,LiD Z,LiQ,et al.Efficient Degradation of Benzene overLaVO4/TiO2Nanocrystalline Heterojunction Photocatalyst under Visible Light Irradiation[J],Environ Sci Technol,2009,43:4 164-4 168.
[30]Chen Y L,LiD Z,Wang X C,et al.H2-O2Atmosphere Increases the Activity of Pt/TiO2for Benzene Photocatalytic Oxidation by Two Orders of Magnitude[J].Chem Commun,2004:2 304-2 305.
[31]Chen Y L,Li D Z,Wang X C,et al.Effects of H2on Photooxidation of Volatile Organic Pollutants over Pt/TiO2[J].New J Chem,2005,29:1 514-1 519.
[32]LiD Z,Che Z X,Chen YL,et al.A New Route forDegradation of Volatile Organic Compounds under Visible Light:Using the Bifunctional Photocatalyst Pt/TiO2-xNxin H2-O2Atmosphere[J].Environ Sci Technol,2008,42:2 130-2 135.
[32]Zhao X,Zhu Y F.Synergetic Degradation of Rhodamine B at a Porous ZnWO4Film Electrode by Combined Electro-Oxidation and Photocatalysis[J].Environmental Science&Technology,2006,40(10):3 367-3 372.
[33]Zhao X,YaoW Q,Wu Y,et al.Fabrication and Photoelectrochemical Properties of Porous ZnWO4Film[J].Journal of Solid State Chem istry,2006,179(8):2 562-2 570.
[34]Zhang S C,Zhang C A,Man Y,et al.Visible-Light-Driven Photocatalyst of Bi2WO6Nanoparticles Prepared Via Amorphous Complex Precursor and Photocatalytic Properties[J].Journal of Solid State Chem istry,2006,179(1):62-69.
[35]Fu H B,ZhangL W,YaoW Q,et al.Photocatalytic Properties of Nanosized Bi2WO6Catalysts Synthesized Via a Hydrothermal Process[J].Applied Catalysis B-Environmental,2006,66(1-2):100-110.
[36]Fu H B,Lin J,ZhangL W,et al.Photocatalytic Activities of a Novel ZnWO4Catalyst Prepared by a Hydrothermal Process[J].Applied Catalysis a-General,2006,306:58-67.
[37]Zhang C,Zhu Y F.Synthesis of Square Bi2WO6Nanoplates as High-Activity Visible-Light-Driven Photocatalysts[J].Chemistry of Materials,2005,17(13):3 537-3 545.
[38]Fu H B,Pan C S,YaoW Q,et al.Visible-Light-Induced Degradation of Rhodamine B by Nanosized Bi2WO6Ρ[J].Journal of Physical Chem istry B,2005,109(47):22 432-22 439.
Progress and Prospects in Research of the Photocatalytic Purification Materials for Aromatic-Polluted Air
CHEN Xun,WANG Xuxu,FU Xianzhi
(Research Institute of Photocatalysis,National Research Center of Environmental Photocatalysis,State Key Laboratory Breeding Base of Photocatalysis,Fuzhou University,Fuzhou 350002,China)
Aromatic hydrocarbon is a kind of volatile,poisonous,and hardly degraded organic compounds.They are very deleterious for environment when evolving in air.Photocatalysis as an advanced oxidation technique is considered asone of the developing directions for environmental purification in the future.However,the classical TiO2material is not only of low activity but also easily deactivated in photocatalytic oxidation of aromatics,which greatly restricts the practical application of photocatalytic technology in the field of air purification.On the basis of a brief review,we summarize our recentwork on novelmaterials and their properties for photocatalytic purification of aromatic-polluted air.The results reveal that there are numerous wide band-gap semiconductor materials with excellent photocatalytic activity and stability that are superior to T iO2.Some materials show very high activity even though under visible light illumination.In addition,it has been found that adding a s mall quantity of hydrogen could greatly increase the photocatalytic efficiency of noblemetal-modified TiO2for benzene degradation,which displays the bright foreground of photocatalytic technique in air purification.
review;materials;photocatalysis;air purification;aromatic hydrocarbon
X131.1
A
1674-3962(2010)01-0010-10
2009-10-19
973计划国家重大基础研究项目(2007CB613306)
付贤智,1957年生,博士,教授,博士生导师
