改进计时电流法的数学模型和电路实现
2010-10-14陈旭海陈敬华潘海波李玉榕林新华
陈旭海 陈敬华 潘海波 李玉榕 杜 民,* 林新华
(1福州大学电气工程与自动化学院,福州 350000; 2福建省医疗器械和医药技术重点实验室,福州 350000;3福建省医科大学药学院药物分析学系,福州 350000)
改进计时电流法的数学模型和电路实现
陈旭海1,2陈敬华3潘海波2李玉榕2杜 民2,*林新华3
(1福州大学电气工程与自动化学院,福州 350000;2福建省医疗器械和医药技术重点实验室,福州 350000;3福建省医科大学药学院药物分析学系,福州 350000)
当采用电化学计时电流法对样品进行检测时,由于检测系统的延迟,采集到的电化学信号会产生偏差,这种现象在多通道电化学信号快速采集时尤为明显.为克服该问题,我们提出一种新的电化学方法来快速产生电流峰值,通过测量峰值快速准确地测出被测物浓度.本文首先为该方法建立理论模型,推导出电流峰值与被测物的浓度关系;再引入经典控制理论中的控制环节来改进传统电化学电路以实现该方法,并加入峰值检测电路来准确获取电流峰值信号;最后,利用该方法研究K3[Fe(CN)6]和3,3′,5,5′-四甲基联苯胺(TMB)溶液的电化学反应,证明了本方法相比传统计时电流法具有更高的信噪比和灵敏度,电流峰值与浓度呈线性关系,并且检测结果不受采样时间延迟的影响,克服了计时电流法的不足.
计时电流法; 电化学分析仪; 峰值检测器; 惯性环节
Abstract:To overcome the disadvantages of chronoamperometry we report a novel electrochemical method where a peak current is quickly generated for the current vs time curve by changing the waveform of voltage excitation in the working electrode.In particular,we derived a mathematical model to illustrate the principle of this method and it can also be used to demonstrate that the peak current is linear with regards to the concentration of the target substance.Moreover,we developed a device with an improved electrochemical circuit using a control element from control theory to change the waveform of voltage excitation.The improved circuit can detect the peak automatically without a precise sample time.Finally,the device was used to study the electrochemical behavior of K3[Fe(CN)6]and 3,3′,5,5′-tetramethylbenzidine(TMB).We show that the method has a better signal to noise ratio and higher sensitivity than chronoamperometry.The obtained peak current is linear with regards to the concentration of the target substance and can be quickly detected without a precise sample time.
Key Words: Chronoamperometry;Electrochemical analyser;Peak detector;Inertial element
计时电流法是一种简单且应用广泛的电化学检测技术.它的工作原理是:在工作电极与参比电极之间施加一个阶跃电势作为激励,由氧化还原反应产生的随时间变化的响应电流流过工作电极和对电极,电流初始值较大,并随时间逐渐减小.Cottrell根据扩散定律,采用拉普拉斯变换对溶液中一个平面电极上浓度的线性扩散做了理论推导,得到了著名的Cottrell方程[1]:

其中,iCottrell为阶跃电势激励下的响应电流;F为法拉第常数;n为电极反应的电子转移数;A为电极表面积;为电活性物质在溶液中的初始浓度;D为电活性物质的扩散系数;t为电解时间.该方程为计时电流法建立了较精确的数学模型,即电流与电解液的本体浓度成正比,且反比于时间的平方根.随后,许多基于计时电流法与Cottrell方程的电化学检测技术迅速发展起来,如:取样电流伏安法、多电势阶跃法、计时电量法等[2-3].由于计时电流法具有硬件要求低、通用性强、稳定性和重复性好等特点,这些基于计时电流法的检测技术亦广泛应用于电化学研究、疾病诊断、环境监测、食品安全等领域[4-8].值得注意的是,近年来基于多通道电化学电极阵列的生物传感器已成为研究热点[9-11],许多研究采用计时电流法,从优化实验条件、改进生物传感器的角度出发,改善多通道电化学生物传感器的灵敏度与特异性[12-14].
然而,计时电流法在实际应用中仍存在一定局限.根据方程(1),电压施加一段时间后,微弱的电流值容易受到仪器内部和外部的电磁干扰,信噪比低.此外,在生化检测中,微量的被测溶液中通常包含生物活性物质,如:细胞、DNA、蛋白质等,这些物质长时间暴露在空气中易失去生物活性,进一步导致信号不稳定,所以实验中不同浓度的被测物采用计时电流法得到的i-t曲线,经过一段时间后往往重叠在一起而难以区分[15-16].因此,施加电压后等待较长时间再进行电流采样来判断被测物浓度,容易导致误差.如果在电压施加后迅速对电流采样,虽然可以实现快速检测,避免干扰,但是由方程(1)可见,在it曲线的初始部分,电流初始值极大,需要检测系统有较宽的测量范围;曲线斜率大,即电流下降快,如果采样时间有微小偏差,就会导致电流采样值的误差,因此需要仪器能精确控制采样时间.然而,在多通道采集系统中,设备需要对多通道生物传感器中的各个电解池依次采样,而设备中的关键元件,如数字/模拟转换器(DAC)、模拟/数字转换器(ADC)、模拟开关等又不可避免地存在一定延迟,所以任意两个电解池的采样时间都不可能完全一样,误差也就难以避免.目前报道的多通道电化学传感器的通道数从几个到上百个[9-11],若相邻两个通道的时间延迟为△t,则设备对第n个通道进行采集时,将会延迟n△t.通道数越多,延迟越严重,误差也就越大.因此,这些缺点在一定程度上限制了基于计时电流法的多通道电化学生物传感器的应用.
这种电流初始值极大且下降迅速的问题主要由工作电极上施加的阶跃电压导致,本文通过以下两步来改进仪器,以实现快速精确的电化学检测:
(1)在传统的电化学恒电势仪中加入控制环节改变激励电压的波形,使得在i-t曲线的初始部分出现峰值,并且电流峰值与被测物的浓度呈线性关系.
(2)在传统的电化学信号检测电路中加入峰值检测器来锁存电流峰值,使得在峰值采样时,无需精确控制采样时间也能获取精确的电流峰值.
通过以上改进,不仅能实现快速精确的电化学检测,由于仪器电路结构简单、对元件性能要求低,所以还可以很方便地扩展为多通道检测电路.本文首先为该方法建立数学模型,证明电流峰值与被测物浓度的关系,然后通过对电路改进实现该方法,最后通过一系列实验来测试该方法.
1 理论部分
1.1 峰值的产生
考虑O+ne→R的体系,其中O是氧化物,R是还原物.传统计时电流法在t=0时刻施加阶跃电势给工作电极,当t>0时,电极表面的氧化物浓度迅速降为0,产生无穷大的电流初始值,并按Cottrell方程的形式随时间迅速递减.实际检测中也可以看到,电流初始峰值极大,往往超出仪器量程且非常接近0点,难以测量.在此,假设为工作电极施加一个较缓慢上升的激励电势,使电极表面氧化物浓度下降变缓,使得在浓度下降和物质传递的过程中出现瞬时的动态平衡,从而产生电流峰值,以便于检测.惯性环节广泛应用于经典控制理论中,并且具有简单可靠的电路结构[17-18],它的特性是:当输入为阶跃信号时,输出缓慢上升,经过一段时间后达到稳态.基于以上思想,我们在恒电势仪的输入端加入一个惯性环节来减缓电压上升的速度,如图1所示,直流输出替代传统电化学电路中的DAC产生阶跃电压,经惯性环节的缓冲作用后再由恒电势仪施加到电解池中的电极上,由此得到的响应电流中将会出现峰值.
以下将通过理论模型来分析峰值产生的机理,并推导出电流峰值与被测物浓度的关系.
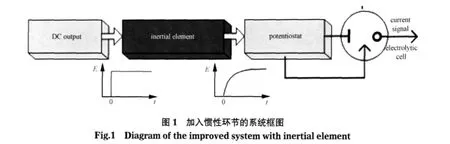
1.2 峰值电流的数学模型
假设在电压施加到工作电极前,即t<0时,被测溶液均匀且氧化物浓度为,而在电压施加的整个过程中,即t>0时,远离电极的本体相不变,氧化物浓度仍为.施加的电压的波形为:

电压E(t)随时间缓慢上升,并趋于稳定值E∞,τ为电路的惯性时间常数,τ越大,电压上升速度越慢.令t时刻,溶液中与电极表面距离为x处的氧化物浓度为CO(x,t),t>0时在电子传递和物质扩散的作用下,假设电极表面的氧化物浓度CO(0,t)以相反的趋势缓慢下降并趋于0,即:

T是电极表面氧化物浓度的惯性时间常数,由电路的惯性时间常数τ决定,τ越大,T亦越大,浓度下降速度越慢.根据Fick第二定律[19],解线性扩散方程:
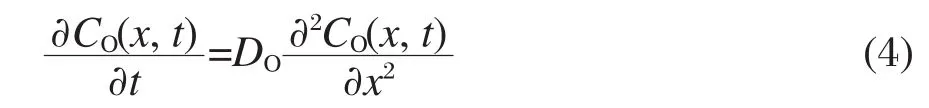
其中DO是溶液中氧化物的扩散系数,设边界条件为:

方程(3)和方程(4)通过拉普拉斯变换为:
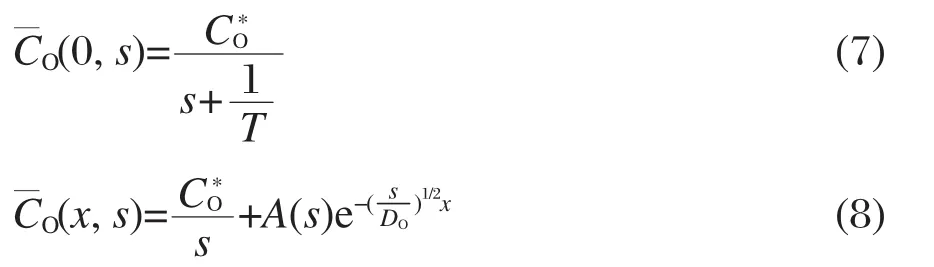
由式(7)和式(8)得:
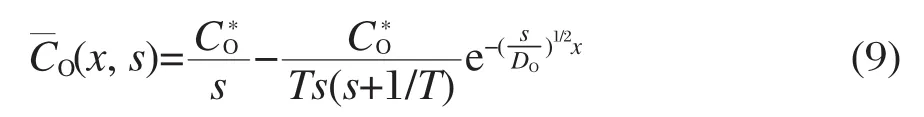
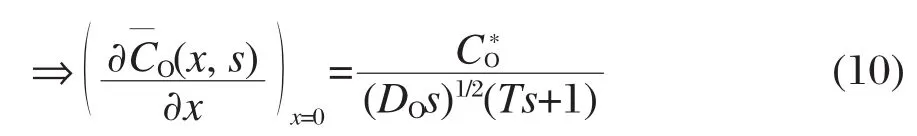
再根据Fick定律[19],可得:

其中JO(0,t)为电极表面氧化物流量,n为电子数,i(t)为流过面积为A的电极表面的电流.将其经拉普拉斯变换得:

将式(10)代入式(12)中,可得电流的拉普拉斯变换为:
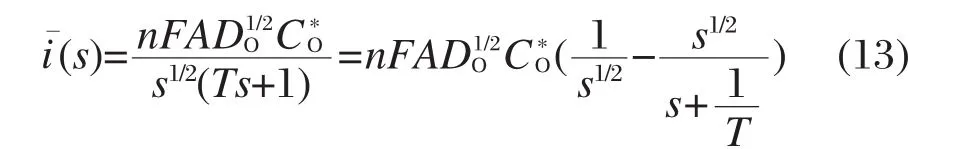
用Matlab对该式进行拉普拉斯反变换,得:

则有:

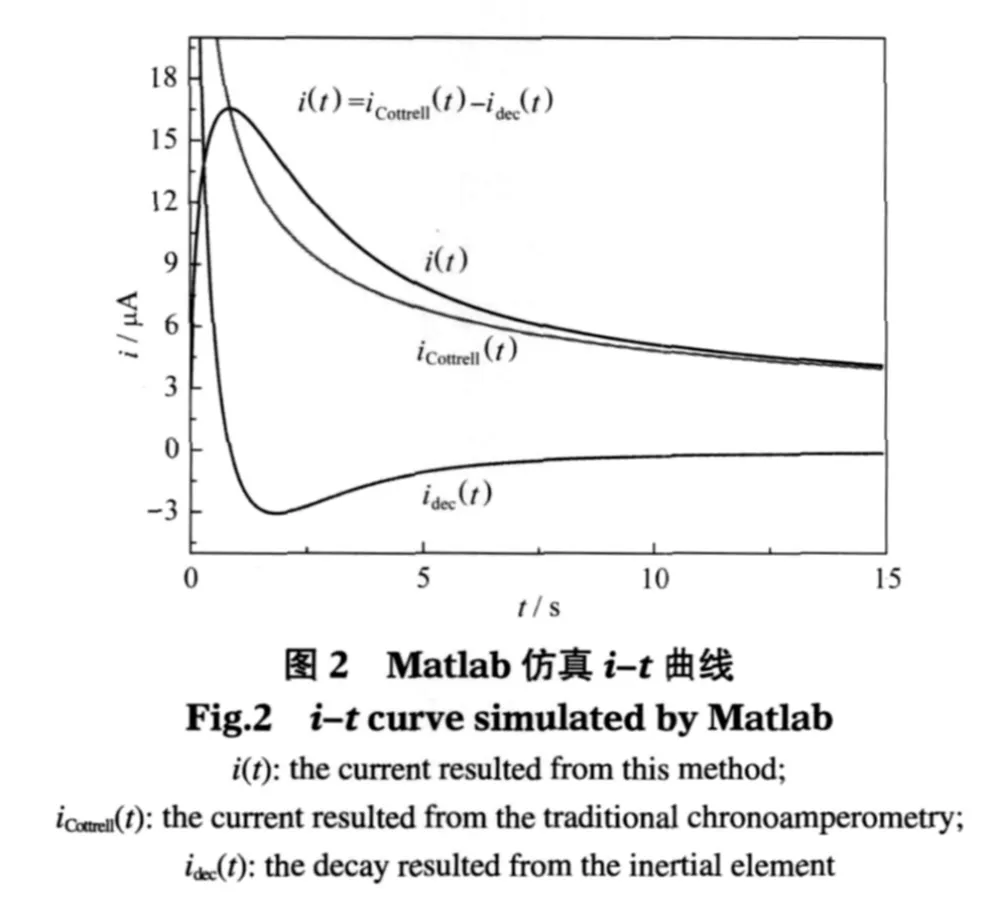
由式(16)可见,电流由两部分组成,第一部分iCottrell(t)为由原始的不含惯性环节的系统产生的电流,第二部分idec(t)可看做由惯性环节所产生的衰减成分.令 n=1,F=9.64853×104C,A=4π×10-6m2,DO=5×10-10m2·s-1,=1 mol·L-1,可得 K=2.7112×10-5.再令T=1 s,并代入式(14),由Matlab绘图得到采用该方法产生的时间电流曲线如图2所示.
由图2可见,在惯性环节的衰减作用下,在t=1s时刻i-t曲线出现峰值.约在 t>10 s后,idec(t)逐渐衰减为0,即惯性环节的衰减作用逐渐消失,此时i(t)曲线逼近iCottrell(t)曲线,因此在曲线的后半段可以用i(t)曲线近似替代iCottrell(t)曲线.
系数K中包含有许多重要参数,如本体浓度、电极表面积等,为求得电流峰值ipeak与K的关系,对式(14)求极值:

由以上分析可见,基于这种方法,我们可以采用峰值检测器自动检测电流峰值,从而快速确定被测对象浓度或电极表面积等参数,且不需要高速的电子器件来精确设定采样时间,大幅降低了仪器成本,尤其适用于多通道检测.以下将通过改进传统电化学电路来实现该方法.
2 实验部分
2.1 电路改进
传统的电化学工作站由恒电势仪、信号采集两个主要部分组成[20-21],目前市场上主流的电化学分析仪和大部分已报道的电化学工作站都具有相似的电路结构[2,22-25],在此不详细叙述传统电化学电路,而着重介绍在传统电路上的改进.改进电化学检测系统的原理框图见图4,其中,inertial element(惯性环节)和peak detector(峰值检测器)是改进后加入的电路模块,其他部分是传统电化学电路的原理框图.
在改进的系统中,加入了一个一阶惯性环节和一个峰值检测器,DAC输出幅值可调的阶跃电压,通过惯性环节转换为上升较缓慢的电压后,再输入到恒电势仪.在实际应用中,可采用电压固定的直流输出模块作为惯性环节的输入,从而进一步简化电路.由电解池产生的电流经过i/V转换器转换为电压,再经滤波放大后输出到峰值检测器,峰值检测器自动保持峰值电压,等待ADC采集.图5(A)为一阶惯性环节电路图,惯性时间常数T由电路中的C2和R6决定,即T=C2×R6.当待测物浓度较高时,可以增大T值来降低峰值电流,从而提高仪器的最高检测限;相反,减小T值,可以提高仪器的灵敏度,从而检测到低浓度的被测物.图5(B)为峰值检测器的电路图,电路由两个运放组成,当输入电压上升时,二极管D6导通,电路相当于电压跟随器,输出跟随输入上升,同时为电容C4充电;当输入电压下降,D6截止,由于运放U7的输入阻抗很大,C4放电电流极小,因此在10 V输入电压的情况下,输出电压在500 ms内可以维持在输入电压的99.99%以上.因此,即使采用低速ADC进行多通道采集,也可以获得精确的峰值电流.

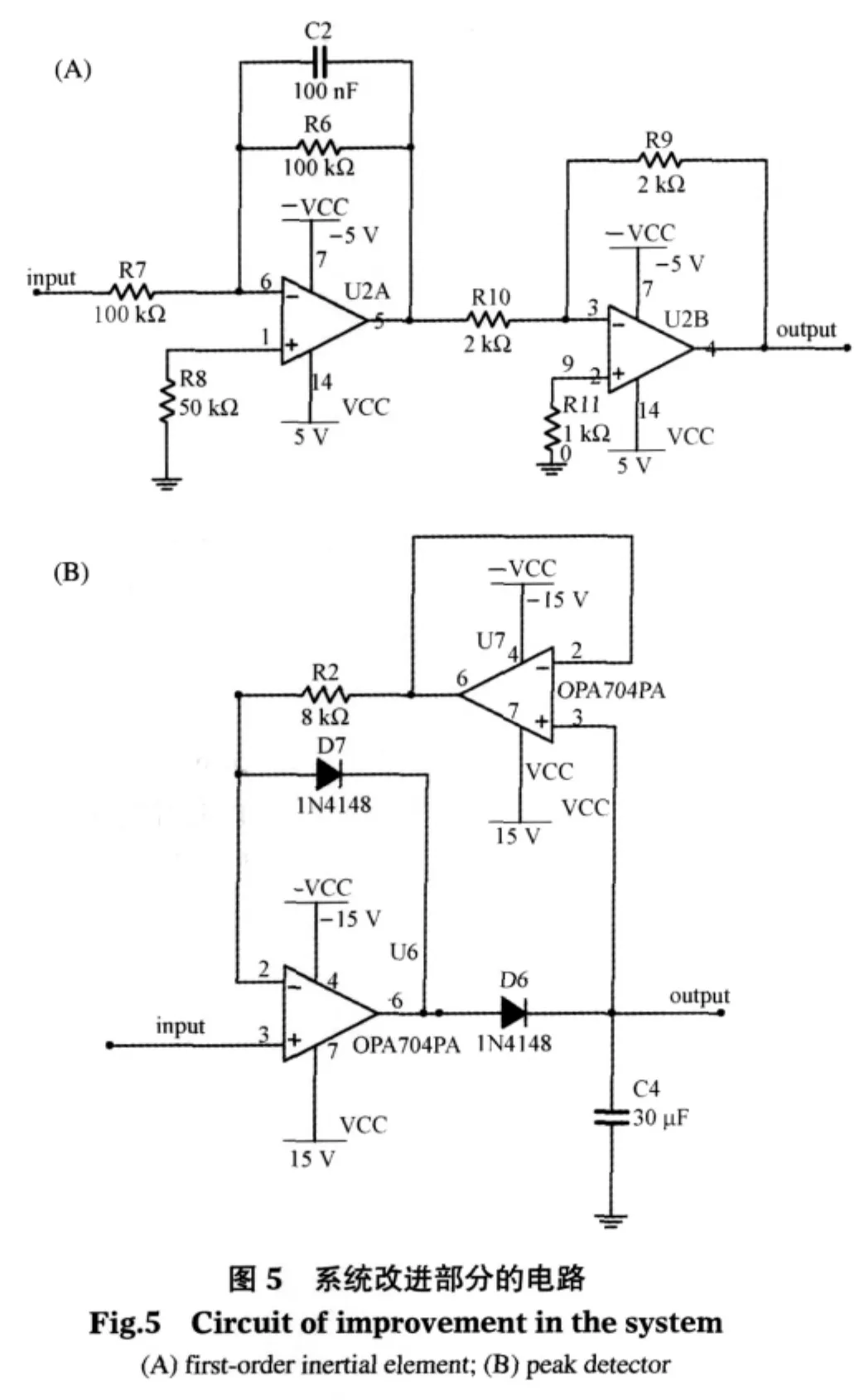
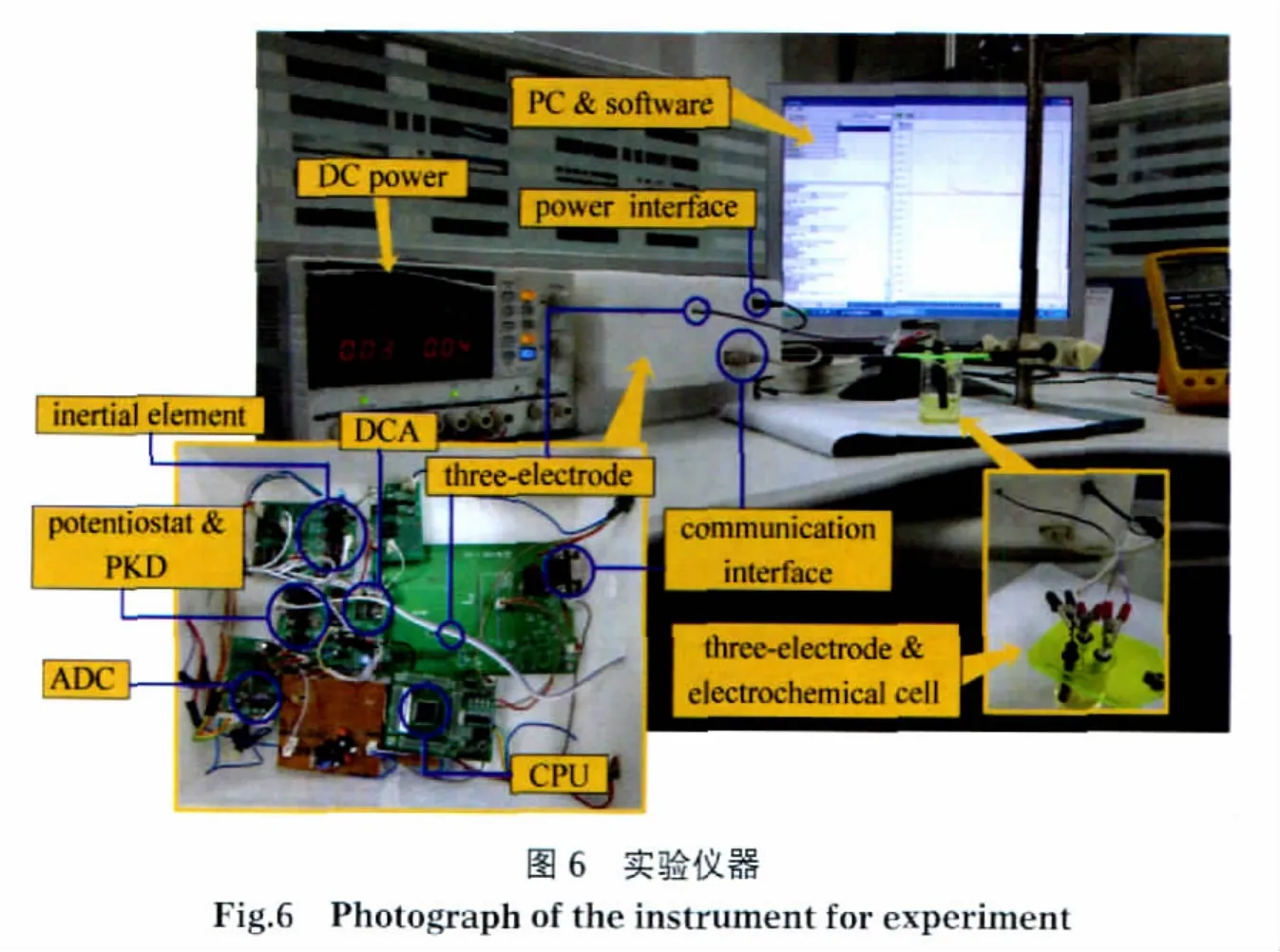
如图6所示,为得到完整准确的信号波形来验证该方法,实验所用的改进后的电化学分析仪由三部分组成:直流可调稳压电源,主机和上位机.采用16位DAC(MAX542)用于输出阶跃电压,16位ADC(MAX1168)用于数据采集,并采用DSP(TMS320 F2806)作为主控制器用于协调仪器各部分硬件和与PC之间的数据通信.如前所述,在实际应用中,由于我们只关心电流峰值大小,可以采用干电池供电、直流稳压电源芯片替代DAC、带ADC的8051核的单片机替代MAX1168和TMS320F2806.该实验仪器采集完整电流波形并通过RS-232接口将数据上传至PC显示和存储,PC机的数据采集软件采用Borland C++builder 6.0编译环境构建,采集到的数据由Matlab7.1做进一步分析处理.
2.2 材料与试剂
K3[Fe(CN)6]购买自美国Sigma公司,TMB底物购买自美国Neogen公司,辣根过氧化酶(HRP)购自北京博奥森生物技术有限公司.HRP酶储备液由牛血清白蛋白(BSA)和HRP按1∶2000的体积比配成指定浓度,并加入灭菌水,4℃保存.所有试剂均由从MilliQ系统制得的超纯水(电阻率:18 MΩ·cm)配制.工作电极为金电极,对电极为铂电极,参比电极为Ag/AgCl电极,均购自上海辰华仪器公司.
3 结果与讨论
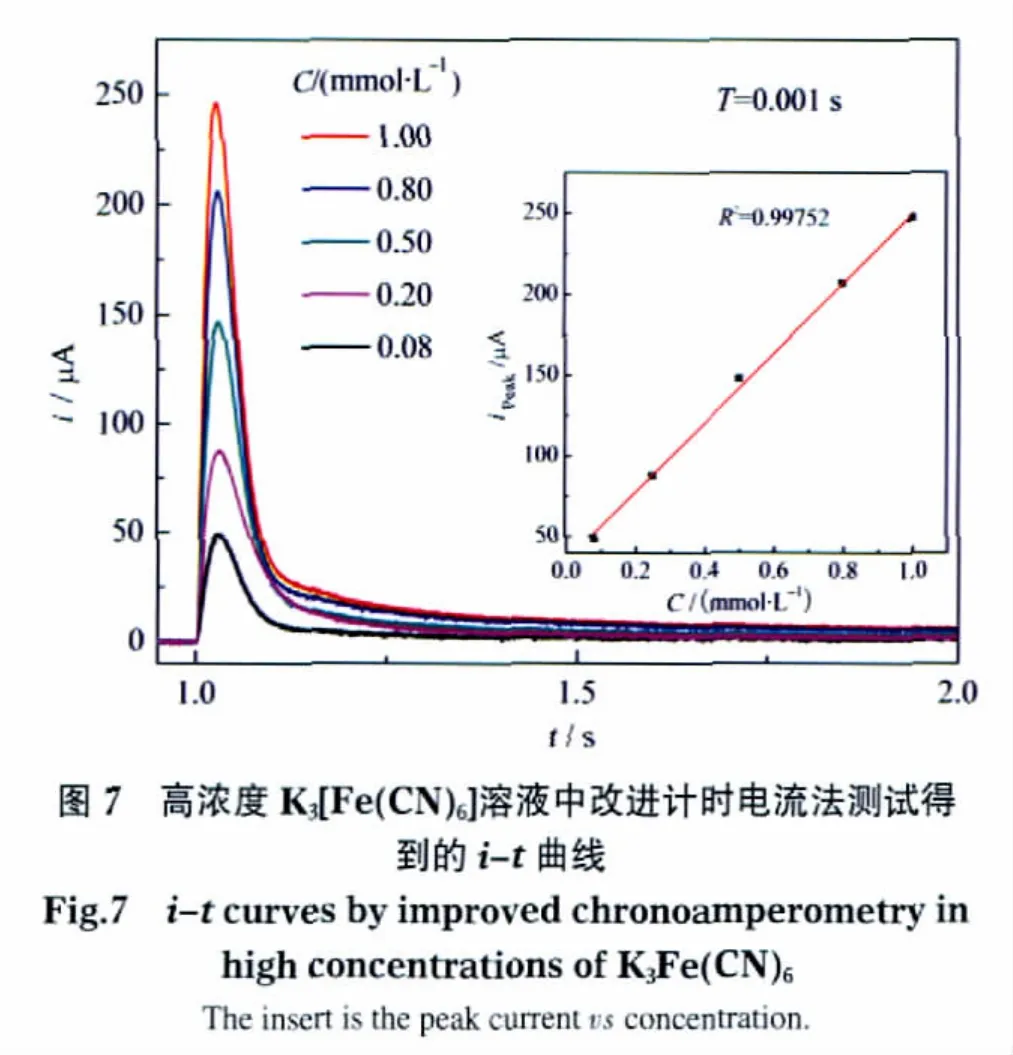
首先,用该装置研究较高浓度的K3[Fe(CN)6]溶液(0.08-1 mmol·L-1)和较低浓度的 K3[Fe(CN)6]溶液(0.625-10 μmol·L-1)的电化学行为.在 t=1 s 时,对较高浓度的K3[Fe(CN)6]溶液施加电压,电压稳定值为0.8 V,如图7所示,在t=1.1 s附近响应电流立刻达到峰值,五次实验后,上位机软件绘制出五条具有不同峰值的曲线,分别对应五种不同浓度的溶液.峰值检测器将峰值长时间保持,等待ADC进行精确采集.由图7的拟合曲线可见,峰值与浓度的线性度极高(线性相关系数为0.99752).同样,尽管浓度较低(图8),电流信号噪声较大,峰电流值仍与溶液浓度保持较高的线性度(线性相关系数为0.99672).但由于电极和溶液接触面存在双电层电容效应,在施加阶跃电压的瞬间,电容因充电而产生微小的峰电流,该电流与被测对象产生的峰电流相叠加,导致图7与图8拟合的直线向上偏移而未通过坐标零点,此外,仪器中运算放大器的零点漂移现象和ADC的基准电压的微小偏差也可能导致该拟合直线未过零点.由于采样值整体偏移,所以在软件或硬件校准时,可以简单地减去一个偏移量来解决该问题.由理论分析可知,由该方法产生的i-t曲线的后段可以近似等同于传统计时电流法产生的i-t曲线,在图7中,1 mmol·L-1的K3[Fe(CN)6]溶液产生的峰电流值是1.8 s时刻电流值的175倍,也就是说,相比传统计时电流法,利用该方法产生的峰电流具有非常高的灵敏度和检测速度,峰电流的信噪比(62 dB)也远高于传统计时电流法(17 dB).尤其在浓度较低的情况下,如图8所示,传统计时电流法得到的电流信号极其微弱,容易受到外界电磁干扰,对应不同浓度的各条i-t曲线的后段相互重叠,而采用该方法得到的峰值电流则具有很高的信噪比和灵敏度.
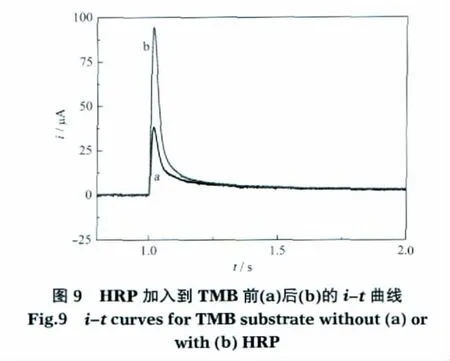
用该装置研究加入微量HRP到TMB-H2O2溶液前后的电化学反应,结果如图9所示.加入3 μL配制好的HRP后,在其催化作用下,TMB被H2O2迅速氧化,电流峰值明显增大.同样,在i-t曲线的后段,两条曲线仍然重叠,没有明显区别,这也再次表明,该方法较传统计时电流法有更高的灵敏度和准确度.
此外,本方法与循环伏安法(CV)相比,具有如下优点:(1)本方法所产生的峰电流值远高于CV法得到的电流峰值,具有更高的灵敏度;(2)采用本方法进行检测,从电压施加到产生峰电流的时间小于1 s,远比CV法检测时间少,更适用于快速检测;(3)本方法所需仪器的硬件结构简单,可用直流输出代替DAC输出,无需高速模拟元件,采用低速ADC即可实现大规模电极阵列芯片的电流峰值采集.但是,相比本方法的电流-时间曲线,以CV法为代表的许多电化学方法记录的是电流-电势曲线,包含了更多的电化学反应的信息,可用于电极反应的机理研究.而本方法则更适用于多通道在现场快速定量检测.未来工作中,本课题组可能将多电势阶跃的思想引入该方法,使其同样适用于电化学反应的机理研究.

4 结 论
本研究报道了一种改进自计时电流法的电化学快速检测方法的理论建模、电路实现,并通过实验证明了该方法较传统计时电流法有更高的灵敏度和准确度,克服了计时电流法的不足.在实际应用中,该装置可以进一步简化,并能很容易地扩展为多通道检测装置.由于该方法通过电流峰值进行定量,不需要精准控制采样时间,因此非常适合于大规模电化学电极阵列的信号检测.
1 Cottrell,F.G.Z.Phys.Chem.,1902,42:385
2 Bard,A.J.;Faulkner,L.R.Electrochemical methods:fundamentals and applications.Trans.Shao,Y.H.;Zhu,G.Y.;Dong,X.D.;Zhang,B.L.Beijing:Chemical Industry Press,2005:111-112,443-445[Bard,A.J.;Faulkner,L.R.电化学方法——原理和应用.邵元华,朱果逸,董献堆,张柏林,译.北京:化学工业出版社,2005:111-112,443-445]
3 Anson,F.C.Anal.Chem.,1966,38:54
4 Ge,F.;Cao,R.G.;Zhu,B.;Li,J.J.;Xu,D.S.Acta Phys.-Chim.Sin.,2010,26(7):1779 [戈 芳,曹瑞国,朱 斌,李经建,徐东升.物理化学学报,2010,26(7):1779]
5 Perez,X.A.;Bianco,L.E.;Andrews,A.M.J.Neurosci Methods,2006,154:245
6 Gasana,E.;Westbroek,P.;Temmerman,E.;Thun,H.P.;Kiekens,P.Anal.Chim.Acta,2003,486:73
7 Micheli,L.;Grecco,R.;Badea,M.;Moscone,D.;Palleschi,G.Biosens.Bioelectron.,2005,21:588
8 Karasinski,J.;White,L.;Zhang,Y.;Wang,E.;Andreescu,S.Biosens.Bioelectron.,2007,22:2643
9 Chu,H.Y.;Kuo,T.Y.;Chang,B.;Lu,S.W.;Chiao,C.C.;Fang,W.Sens.Actuators A,2006,130:254
10 Williams,G.;D′Silva,C.Sens.Actuators B,1996,30:151
11 Yun,Y.H.;Dong,Z.Y.;Shanov,V.N.;Doepke,A.;Heineman,W.R.;Halsall,H.B.;Bhattacharya,A.;Wong,D.K.Y.;Schulz,M.J.Sens.Actuators B,2008,133:208
12 Wei,F.;Wang,J.H.;Liao,W.;Zimmermann,B.G.;Wong,D.T.;Ho,C.M.Nucleic Acids Res.,2008,36:e65
13 Liu,G.;Wan,Y.;Gau,V.;Zhang,J.;Wang,L.H.;Song,S.P.;Fan,C.H.J.Am.Chem.Soc.,2008,130:6820
14 Wang,K.;Chen,J.H.;Chen,J.;Liu,A.;Li,G.W.;Luo,H.B.;Lin,X.H.;Chen,Y.Z.Electroanalysis,2009,21:1159
15 Jeon,M.K.;Zhang,Y.;McGinn,P.J.Electrochim.Acta,2009,54:2837
16 Santasalo,A.;Vidal-Iglesias,F.J.;Solla-Gullón,J.;Berná,A.;Kallio,T.;Feliu,J.M.Electrochim.Acta,2009,54:6576
17 Jumarie,G.Robotica,1990,8:73
18 Han,Z.X.;Sun,Y.Proceedings of the Chinese Society for Electrical Engineering,2002,22(4):118 [韩忠旭,孙 颖.中国电机工程学报,2002,22(4):118]
19 Fick,A.Poggendorff′s Ann.Physik.,1855,94:59
20 Kissinger,P.T.;Heineman,W.R.Laboratory techniques in electroanalytical chemistry.New York:Marcel dekker,1996:165
21 Schwarz,W.M.;Shain,I.Anal.Chem.,1963,35:1770
22 Jin,Y.;Wang,H.;Lv,Z.L.;Yang,S.Y.;Cai,H.Y.;Jiang,J.F.Tsinghua Science and Technology,2009,14:593
23 Huang,C.Y.;Tsai,T.C.;Thomas,J.L.;Lee,M.H.;Liu,B.D.;Lin,H.Y.Biosens.Bioelectron.,2009,24:2611
24 Serra,P.A.;Rocchitta,G.;Bazzu,G.;Manca,A.;Puggioni,G.M.;Lowry,J.P.;O′Neill,R.D.Sens.Actuators B,2007,122:118
25 Reay,R.J.;Flannery,A.F.;Storment,C.W.;Kounaves,S.P.;Kovacs,G.T.A.Sens.Actuators B,1996,34:450
Mathematical Model and Circuit Realization to Improve Chronoamperometry
CHEN Xu-Hai1,2CHEN Jing-Hua3PAN Hai-Bo2LI Yu-Rong2DU Min2,*LIN Xin-Hua3
(1College of Electrical Engineering and Automation,Fuzhou University,Fuzhou 350000,P.R.China;2Fujian Key Laboratory of Medical Instrumentation&Pharmaceutical Technology,Fuzhou 350000,P.R.China;3Department of Pharmaceutical Analysis,Faculty of Pharmacy,Fujian Medical University,Fuzhou 350000,P.R.China)
O646
Received:May 27,2010;Revised:July 27,2010;Published on Web:September 23,2010.
*Corresponding author.Email:fjkeylab@163.com;Tel:+86-591-83759450.
The project was supported by the National High Technology Research and Development Program of China(863)(2008AA02Z433,2006AA02Z4Z1),
International Science and Technology Cooperation Project of China(2009DFA32050)and Natural Science Foundation of Fujian Province,China
(2008J1005).
国家高技术研究发展规划项目(863)(2008AA02Z433,2006AA02Z4Z1),科技部国际合作项目(2009DFA32050)及福建省科技厅重点项目(2008J1005)资助
