有机电化学法合成二茂镍的研究
2010-09-15乔庆东
李 琪, 乔庆东
(辽宁石油化工大学石油化工学院,辽宁抚顺 113001)
有机电化学法合成二茂镍的研究
李 琪, 乔庆东*
(辽宁石油化工大学石油化工学院,辽宁抚顺 113001)
对环戊二烯的提纯和二茂镍的电化学合成过程进行研究,以提纯后的环戊二烯为原料,金属镍为阳极和阴极,溴化钠作导电盐,N,N二甲基甲酰胺(DM F)作有机溶剂,采用氮气保护,恒流源电解,合成了二茂镍。结果表明,双环戊二烯的最佳解聚温度170℃,环戊二烯提纯时的最佳回流温度120℃,且提纯过程必须连续,用气相色谱法检测双环戊二烯和环戊二烯的纯度分别为95.4%和97.3%。在电合成实验中,首先以甘汞电极为参比电极跟踪电解合成过程的电极电位变化,并作极化曲线图,确定最佳电流密度3.93 m A/cm2。其次,通过观察溶液的颜色变化、电极电位的变化和反应溶液的紫外可见、红外谱图变化确定实验最佳反应时间为210 min。最后,通过镍板的减少量计算电流效率为82.87%。
二茂镍; 双环戊二烯; 环戊二烯; 极化曲线
二茂镍,又称二环戊二烯基镍,CAS编号1271 -28-9,分子式C10H10Ni,相对分子质量188.90,密度1.47 g/cm3,为深绿色针状晶体,熔点为172~ 173℃(在抽真空封闭管中,130℃升华)。由于在分子轨道的反键轨道上有填充电子,致使其结构不稳定,遇空气和光缓慢分解,被氧化后变为橙黄色C10H10Ni+[1]。二茂镍不溶于水,溶于乙醚、苯、四氢呋喃等有机溶剂中,但溶解在丙酮、乙醚、乙醇中会分解[2]。单环戊二烯镍和双环戊二烯基镍,可作为烃类氰代、取代衍生物的基本原料,烃类精炼催化剂、氢化催化剂、自由基聚合反应剂、硫化加速剂、燃料抗震剂,亦用于镀镍、制备高纯镍、炔烃聚合的催化剂等[3-6]。关于二茂镍的化学合成方法可见文献[7],与二茂镍电合成相似的是二茂铁的电合成[8-9]。
本文采用的原料是环戊二烯,它通常是以双环戊二烯的形式存在和储存[10],可以制备许多有用环戊二烯金属配合物[11],但关于二茂镍的电合成还未见报到。实验以电化学方法合成了二茂镍,获得了许多有价值的数据。
1 实验部分
1.1 试剂及仪器
试剂:工业双环戊二烯(质量分数为80%,抚顺裕龙化工有限公司),其它试剂均为分析纯。
仪器:YJ/5型直流稳流电源(北京电表厂); 7890Ⅱ型毛细管柱气相色谱仪(上海天美科学仪器有限公司);PXD-2型通用离子计(国营江苏电分析仪器厂);722型紫外可见分光光度计(上海市光学仪器厂);TJ270—30型红外分光光度计(天津市光学仪器厂)。
1.2 原料的制备和提纯
将粗双环戊二烯进行常压蒸馏,切割出170℃(液相温度)以下的馏分。取该馏分再进行回流,回流时要加入适当的铁粉和沸石,回流过程中需要缓慢加热,待温度上升至120℃时停止加热,此时只有极少量的回流现象。经气相色谱检测实验中的双环戊二烯质量分数为95.4%。将回流后的溶液加热至170℃进行解聚反应,在此温度以下的馏分蒸出,此时溶液的温度会保持在170℃不变化,收集气相温度为40~45℃的馏分,即为环戊二烯。由于环戊二烯在常温下能发生二聚反应,所以直接滴入提纯后的N,N-二甲基甲酰胺中备用。
金属镍片使用前要除油除锈,即先将镍片放入质量分数为10%的NaOH中,浸泡30 m in除去镍板表面的油层,取出后用去离子水冲洗,然后将其浸泡在体积分数为10%的盐酸中30 min除去镍板表面的氧化层。取出后先用去离子水清洗3~5次,自然放置干燥。
1.3 电化学合成实验
准确称取作为正极的镍板的质量,组装好反应装置。取实验制备的环戊二烯10 m L,滴入到60 mL提纯后的DM F中,加入1 g导电盐NaBr,迅速密闭反应装置,通入N2以除去空气,通过磁力搅拌加速导电盐的溶解。20 min后导电盐全部溶解,接通恒流电源,反应时间为210 min,最终的溶液为暗绿色,密闭保存于棕色瓶中。迅速从该瓶中取出10 mL溶液倒入20mL冰盐酸中,会有大量的沉淀物出现,抽滤,并将该沉淀物放入石油醚中重结晶,在石油醚溶液中有暗绿色的晶体出现,再抽滤,得暗绿色晶体,即为二茂镍产品,密闭保存。极化曲线的测定是在电解反应时进行的,以甘汞电极为参比电极,用盐桥和PXD-2型通用离子计,在搅拌下测量阳极的电极电位。
2 结果与讨论
2.1 环戊二烯的提纯
环戊二烯在常温下极易产生二聚体,而二聚体在150℃时分解为环戊二烯,170℃时分解速度为30 mo l/h,利用这一特性可提纯得到环戊二烯,反应方程式如式(1)所示:
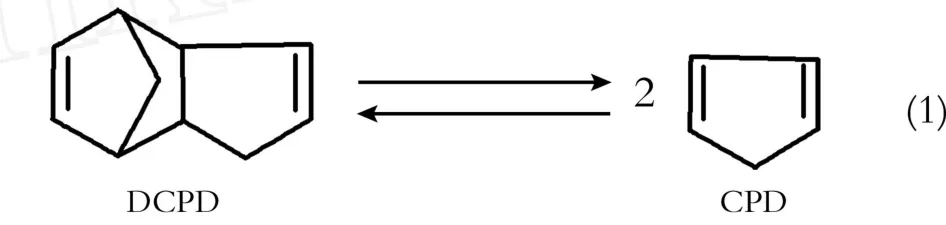
首先将工业双环戊二烯进行蒸馏,收集120℃时的馏分,经气相色谱检测双环戊二烯(DCPD)质量分数为95.4%。将该馏分加热至170℃进行解聚反应,并经再次蒸馏,收集40~45℃的馏分,即为环戊二烯(CPD),其纯度可达97.3%,产品直接滴入提纯后的N,N-二甲基甲酰胺中备用。原料双环戊二烯和环戊二烯的色谱图如图1所示。
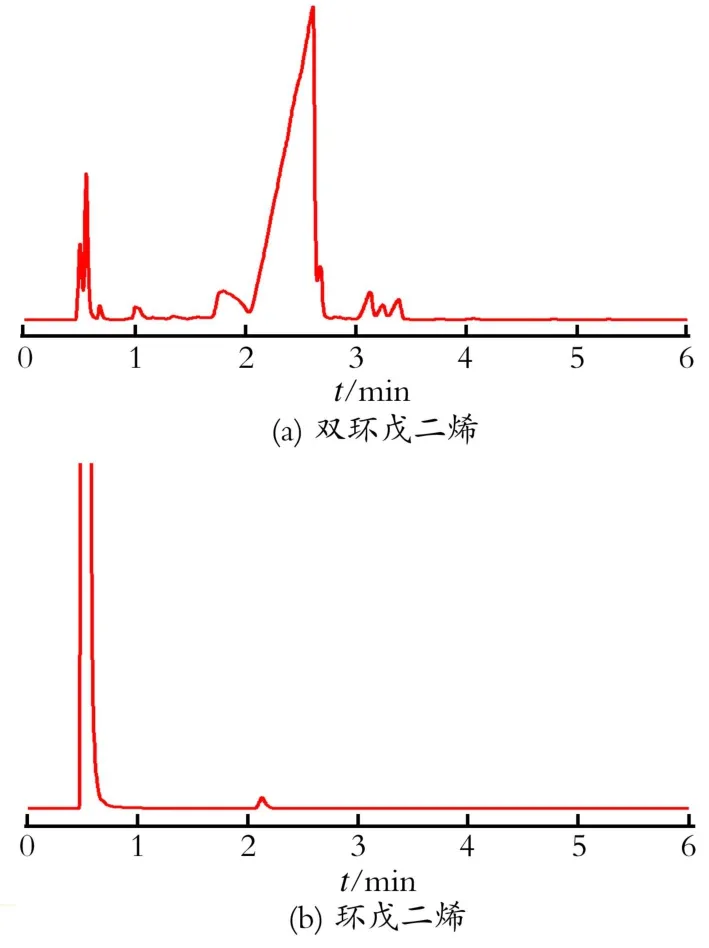
Fig.1 Gas chromatogram of raw dicyclopentadiene and cyclopen tadiene图1 原料双环戊二烯和环戊二烯气相色谱图
在双环戊二烯的加热解聚实验中,152℃时有大量的馏分流出,温度缓慢上升170℃时稳定,此时双环戊二烯开始解聚为环戊二烯。在解聚过程中,新蒸馏的产品与放置一段时间的产品回流时,回流时间的长短有很大的差别。前者的回流时间一般在2~3 h,但后者只需20 min,二者的回流温度变化也很显著,前者变化速度较慢,尤其是42~60℃需要2 h,过60℃以后温度上升较快。而后者的温度几乎是直线上升,如果温度控制不当,就会超过120℃。经过电合成实验后发现,放置一段时间的解聚产品参加的电化学反应的现象不稳定。由此说明,放置后的解聚产品,在空气中已经被氧化或二聚,纯度下降,所以在进行电化学反应前制取的环戊二烯反应一定要连续,并将其直接滴入到DM F中以稀释CPD的浓度,尽量减少氧化或二聚反应的发生。
2.2 电流效率的测算
在直流电的作用下,反应体系中的环戊二烯直接与镍反应生成二茂镍。电解开始时,电解质中的Na+向阴极转移,在阴极上被还原并与环戊二烯反应生成环戊二烯基钠和氢气。与此同时阳极上的Ni被氧化成Ni2+并向阴极转移,与阴极上的环戊二烯基钠生成二茂镍,释放出Na+。电极反应如下:
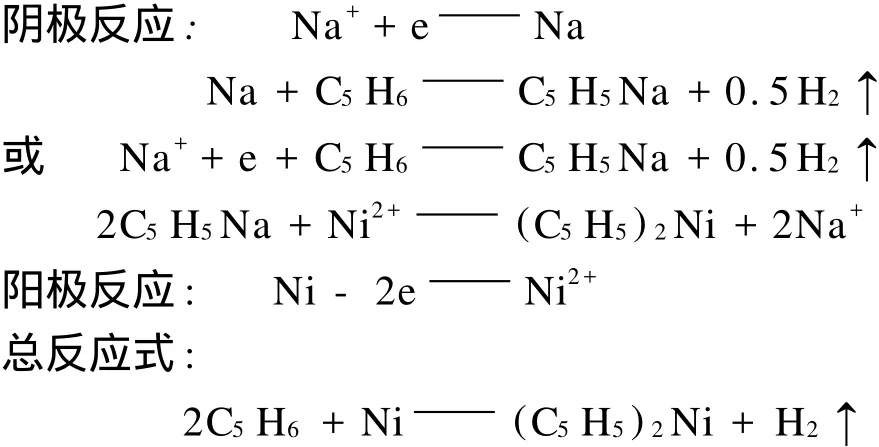
电合成的时间可按Faraday定律来计算:环戊二烯密度0.805 g/m L,摩尔质量66.1 g/mol,实验中环戊二烯的物质的量为0.121 8 mol,反应所需的电荷总量为1.175×104C;当电解反应恒流为44 mA时,环戊二烯完全反应所需要的时间为74.2 h。电流效率是以电解反应前后镍板的质量变化量来计算的。因为电化学反应(Ni-2e=Ni2+)的电流为44 m A,电解时间3.5 h,故电荷总量为554.4 C。而实验测定镍板的质量减少量为0.14 g,对应的电荷量为459.45 C,所以电流效率为82.87%。
2.3 导电盐的选择
导电盐不仅仅起到导电的作用,在此实验中亦起到催化剂的作用。因此,导电盐的选择是十分重要的。阳极为金属Ni,阴极可以使用对电解质惰性的导电物质,如A l、Hg、Pb、Zn、Ni等,该实验必须在严格无水条件下进行,所用试剂应预先干燥。同时电流要严格控制,如果电流过大就会产生胶体。
首先使用溶解度较小NaCl的作为导电盐。但NaCl的溶解速度很慢,电解电压达到50 V时,电流才2.8 m A,故NaCl不是理想的导电盐。当向同样的反应体系中加入同质量的溴化钠时,则溶解速度很快,而且当电压达到3.2 V时电流值就可达到60 mA,所以溴化钠可作为本实验的导电盐。
2.4 保护方式的选择
实验初始是在空气中进行,结果发现,反应进行10 m in左右,溶液就变为绿色,但经过1.5 h后溶液就变为黄色,这是由于二茂镍溶解在有机溶剂中很容易被氧化变质所致[3]。所以电合成实验必须在隔绝空气条件下进行。首先用液封的方法,由于DM F不与饱和烃相溶,在实验中选择了密度比DM F小的石油醚作为液封溶液。虽然石油醚与DM F和CPD的界面不是十分明显,但仍然存在。电解时,在磁力搅拌下液液界面很快消失,但反应混合溶液很快变为绿色,1.5 h后溶液的颜色变黄,这可能是生成的二茂镍已经被氧化,说明该方法不可行。最后使用氮气作为保护气体,反应溶液的颜色始终为暗绿色,可进行电解实验,表明氮气保护作用最好。
2.5 极化曲线
电解反应是在恒电流条件下进行,电解液的极化曲线如图2所示。从图2中可以看出,电极电位随电流的增加而增加较快,后来逐渐放缓,有一段几乎没有太大的变化,稳定值于-1 700 m V左右。如果再增加电流,电位会继续增加,说明有副反应产生。为了保证反应有较高的电流效率,取电极电位稳定值时的较高电流44 m A或3.93 m A/cm2,并将该电流值定为最佳反应电流应用到电合成实验中。
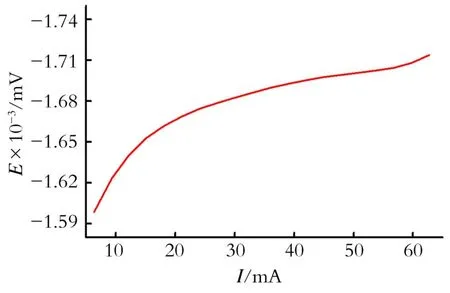
Fig.2 Polarization curve of the reacting solution图2 电解液的极化曲线
2.6 反应时间的选择
二茂镍的反应流程如图3所示[12]。从图3中可看出,反应时间不宜过长或过短,如果时间控制不合理就会有多种副产物生成。实验时可观察到反应前的溶液是无色澄清溶液,随着反应的进行,溶液的颜色由无色变为浅绿,再变为暗绿,但当反应进行到4.5 h左右时反应溶液的颜色开始逐渐变黄,证明此时副反应已经发生。这由于在电解合成的过程中,随着二茂镍不断溶解于有机溶剂中,其浓度不断增加,当浓度达到一定值时,就发生了副反应。根据溶液的颜色变化,副反应发生在阳极,产物可能是(C5H5)2Ni+,反应方程式如式(2)所示:
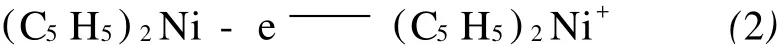
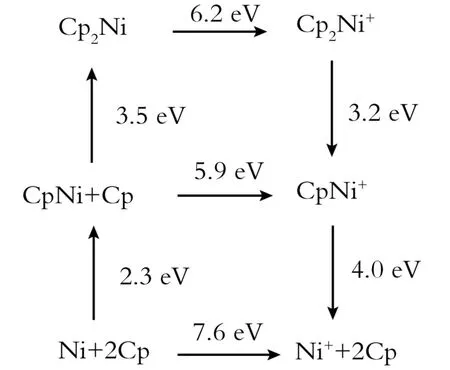
Fig.3 Reaction scheme of n ickelocene图3 二茂镍的反应流程
根据溶液的颜色和电位的变化,以及反应溶液的紫外光谱检测,每隔30~60 min通过导管吸取2 mL反应溶液测定紫外可见光谱图,观察谱图中生成物和反应物在反应溶液中的浓度变化,以二茂镍的浓度最大值为终止时间,表1为不同时间的产物在320 nm处最高吸收峰高值。由表1可知,当反应时间为210 m in时,有最高的吸收峰值,说明此时产物的浓度最高。因此,最佳时间为210 min。
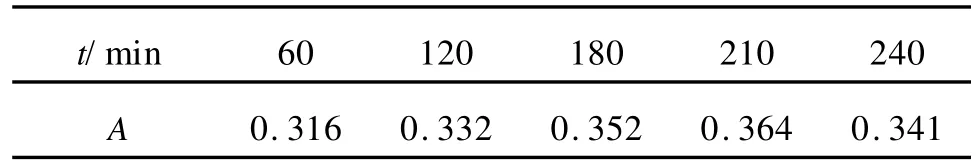
表1 不同时间的产物在320 nm处最高吸收峰高值Table 1 The peak height at 320 nm of production at different time
2.7 产物分析
产物紫外光谱的最大吸收波长为320 nm,而溶剂DM F的最大吸收波长273 nm,故溶剂不影响产物的测定,可直接进行产物的测定。图4是反应开始前和开始60 min后的产物混合液紫外光谱。
图5是溶剂和产物的红外光谱。实际上,在分子在振动过程中,只有偶极矩发生变化的振动方式才能吸收红外光,并在红外光谱中出现吸收带,这种振动方式称为红外活性[13]。反之,在振动过程中,偶极矩不发生变化的振动方式是非红外活性的,虽然有分子振动,但不能吸收红外光。一般的极性分子具有偶极矩,非极性分子不具有偶极矩。

Fig.4 Ultraviolet spectra of the product图4 产品紫外光谱
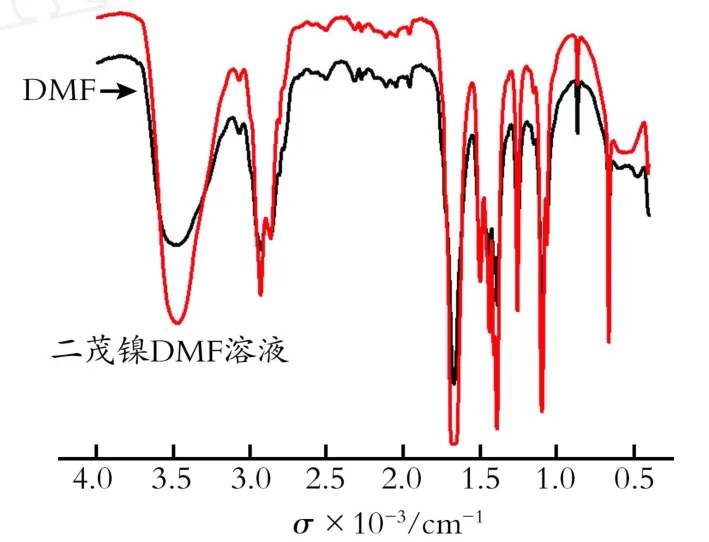
Fig.5 Infrared spectra of DMF and n ickelocene DM F solution图5 DM F和二茂镍DM F溶液红外光谱
由图5可知,产物的谱图中并没有产生其他特征吸收峰,只是某些位置上的峰高度有所变化。因为峰高的大小代表溶液中所含该物质的量的多少,经比较得出,代表—CH和—CH3基团的峰高都有所增强。因此,推断是在电解的过程中有一部分环戊二烯开环,增加了溶液中—CH和—CH3基团的含量,导致峰高增加。而二茂镍的非红外活性使得它在谱图中没有表现出特征吸收峰,与此同时,也没有出现其他物质的特征吸收峰,推测是生成的二茂镍含量很高。
[1] Elschenbroich C.O rganometallics[M].Weinheim:Wiley-VCH,2006.
[2] 陶文田,黎心懿.现代化学试剂手册(第五分册金属有机试剂)[M].北京:化学工业出版社,1992:776.
[3] 《化学化工大辞典》编委会.化学化工大辞典(上)[M].北京:化学工业出版社,2003:559.
[4] Pasynkiew icz S,Pietrzykow ski A,Bukow ska L,et al.A lkylation of cyclopentadienyl rings in the reactionsof nickelocene w ith organolithium compounds[J].Journal of organometallic chemistry,1999,585(2):308-314.
[5] Pasynkiew icz S,Buchow icz W,Popaw ska J,et al.Reactions of nickelocene w ith methyl-,ethyl-and vinyl-lithium compounds,hydrogen elimination and cyclopentadienyl ring hydrogenation[J].Journal of o rganometallic chemistry, 1995,493(1-2):189-195.
[6] Pasynkiew icz S,Oledzka E,Pietrzykow ski A.Polymerization of alkynes on nickelocene based catalysts:considerations on polymerization mechanism[J].Journal of molecular catalysis A:Chemical,2004,224(1-2):117-124.
[7] Wilkinson G,Pauson P L,Birmingham J M,et al.Synthesis of nickelocene[J].Journal of the American chemical society,1953(75):1011-1013.
[8] 杨亚立.有机电化学法合成二茂铁[J].大庆高等专科学校学报,2001,21(4):71-72.
[9] 乔庆东,李琪,孙悦.二茂铁的电合成[J].化工科技,2007,15(2):14-16,27.
[10] 杨春育,丁明,晁建平,等.由环戊二烯合成降冰片烯的动力学[J].石油化工高等学校学报,1999,12(3):35-38.
[11] 刘俊华,曹祖宾,赵德智,等.汽油抗爆剂的研究进展[J].辽宁石油化工大学学报,2004,24(3):48-52.
[12] Sergey Y K,Heinrich L S,Edward W S,et al.M ultiphoton ionization of jet-cooled nickelocene w ith tunable nanosecond laser pulses[J].Chemical physics,2003(293):91-97.
[13] 张旭之,马润宇,王松汉,等.碳四碳五烯烃工学[M].北京:化学工业出版社,1998:625-626.
(Ed.:SGL,Z)
The O rganic Electrochemical Synthesis of Nickelocene
L IQi,Q IAO Qing-dong*
(School of Petrochem ical Engineering,L iaoning Shihua University,Fushun L iaoning 113001,P.R.China)
19 A pril 2010;revised 13 M ay 2010;accepted 24 June 2010
The purification of cyclopentadiene and the electrosynthesis of nickelocene were studied.Nickelocene was electrochemical synthesized by using purified cyclopentadiene as the raw material,and metal nickel used as both anode and cathode,sodium bromide as the conducting salt,N,N-dimethylformamide(DM F)as the o rganic solvent,and nitrogen as the p rotection gas of the solution.In the dicyclopentadiene purification experiment,the best depolymerization and best circum fluence temperature of cyclopentadiene are 170℃and 120℃respectively.The purification p rocess above must be continuously carried on.The purities of dicyclopentadiene and cyclopentadiene determined by gas chromatography are 95.4% and 97.3%.In the electrochemical synthesis experiment,the electrode potential changes are first determined using calomel electrode as the reference electrode;meanw hile the polarizing curves are also measured in order to confirm the best current density,i.e.3.93 m A/cm2.Secondly the best reaction time determined is 210 min through the observation of the solution colo r changes,the electrode po tential changes,the ultraviolet and the infrared spectrogram of the solution.Finally the current efficiency is 82.87%by calculating themass decrement of nickel board during the experiment.
Nickelocene;Dicyclopentadiene;Cyclopentadiene;Polarizating curve
O646.542;TQ151.4
A
10.3696/j.issn.1006-396X.2010.03.014
1006-396X(2010)03-0058-04
2010-04-19
李琪(1969-),女,辽宁抚顺市,副教授,在读硕士。
辽宁省自然科学基金项目(20082187);辽宁省教育厅科技攻关项目(2008379)。
*通讯联系人。
*Co rresponding author.Tel.:+86-413-6860858;fax:+86-413-6860858;e-mail:qiaoqingdong@163.com
