乙炔与金属氯化物络合作用的第一性原理研究
2010-09-15陈永昌孙兆林
李 强, 姜 恒, 丁 勇, 陈永昌, 孙兆林*
(1.辽宁石油化工大学辽宁省石油化工重点实验室,辽宁抚顺 113001; 2.兰州大学化学化工学院,甘肃兰州 730000)
乙炔与金属氯化物络合作用的第一性原理研究
李 强1, 姜 恒1, 丁 勇2, 陈永昌2, 孙兆林1*
(1.辽宁石油化工大学辽宁省石油化工重点实验室,辽宁抚顺 113001; 2.兰州大学化学化工学院,甘肃兰州 730000)
采用密度泛函(DFT)计算方法,优化了不同金属氯化物与乙炔络合的稳定几何构型,并对此络合物体系的电子结构进行了计算和全面分析研究。结果表明,与乙炔吸附络合后,金属氯化物对乙炔的电子有吸引作用,使得乙炔碳碳叁键和金属原子之间产生离子性相互作用,由此乙炔得到活化。计算得到了乙炔几何构型、吸附能和电子变化与金属Mayer价指数的关系,并探讨了不同金属对乙炔活化的可能性大小,为乙炔法合成氯乙烯非汞催化剂的研发提供了理论基础。
乙炔; 金属氯化物; 络合; 第一性原理
A rmentrout PB等[1]科学家自20世纪80年代开始便对过渡金属阳离子与小分子的作用进行过研究[2-5]。过渡金属阳离子与小分子作用时,由于不同金属的电负性不同,所以对小分子的吸附行为也有不同。众所周知,过渡金属因电子组态的不同,其化学性质会有很大的差异。并且当过渡金属与底物分子作用时,其电子组态会发生变化,这正是过渡金属具有催化性能的本质所在。此外,分子体系电子组态的变化与分子内电子的行为密切相关。因此,准确测定过渡金属络合物分子的电子行为至关重要,而实验上对电子结构最直接的表现为测定过渡金属络合物分子的振动[6-8]。但是,小分子络合吸附作用的模拟计算较少,而且小分子在不同金属上吸附作用的系统归纳分析也比较罕见[9]。
乙炔法合成氯乙烯非汞催化剂的研发历年来都受到了国内外学者的关注[10-11]。目前该反应体系普遍使用的催化剂是氯化汞,公认的催化反应机理认为乙炔和氯化氢的催化反应过程首先是乙炔和催化剂生成中间化合物—氯乙烯金属氯化物[12],而此反应中乙炔与催化剂的吸附络合作用决定着该反应的进行难易。乙炔氢氯化法催化剂的活性与金属的电负性有关,电负性越大的金属离子在反应中电子的变化也越多,这种变化可以体现在Mayer价指数的变化,所以讨论乙炔吸附在金属氯化物前后金属离子M ayer价指数的变化可以作为不同金属离子的比较因素。
本研究利用密度泛函理论计算不同金属氯化物对乙炔的吸附作用,通过对吸附体系前后金属氯化物和乙炔的几何、能量和电子结构的变化进行分析对比,从一个全新的角度预测各种金属氯化物作为催化剂对乙炔氢氯化反应的影响。
1 计算方法
选取各种金属的氯化物单体作为研究的对象,使各种金属氯化物和单个乙炔分子发生吸附络合作用。运用密度泛函理论优化并求得金属氯化物及其与乙炔发生络合后乙炔和金属氯化物的结构参数、能量和电子结构数据。
理论计算平台是应用美国Accelrys公司开发的Materials Studio 3.1软件包。DFT计算采用Dmol3模块。DFT计算参数设置如下:计算方法为GGA,选用BP函数,基组采用可极化的双数基组(DNP),中心电子的处理使用全电子计算,系统自旋状态为Spin-unrestricted,自洽场(SCF)参数的建立使总能量收敛至1×10-5Ha。
2 结果与讨论
2.1 吸附络合的结构及吸附能
研究计算了Cu+、Ag+、Hg2+、Cd2+、Zn2+、Pd2+、Cu2+、Ni2+、M n2+、Ca2+、M g2+、Pb2+、Au3+、A l3+等离子形成的氯化物,并计算乙炔在这些氯化物上的吸附,得到优化后的吸附络合结构如图1所示。其中,乙炔在二价金属氯化物上的吸附络合模式主要有两种:一种是乙炔与金属氯化物共面,如CaCl2、CdCl2、HgCl2等;另一种是乙炔与金属氯化物异面垂直,如NiCl2、PdCl2。

Fig.1 Adsorption geometry of acetylene on themetal chlorides图1 金属氯化物吸附乙炔的几何结构
由于金属离子的作用,乙炔的几何结构会发生变化。表1为各金属离子与乙炔吸附前后的吸附能及结构参数。由表1可知,吸附后乙炔的碳碳叁键键长都略有增加,且金属的价态变化多少对碳碳叁键键长的增加有显著的影响。
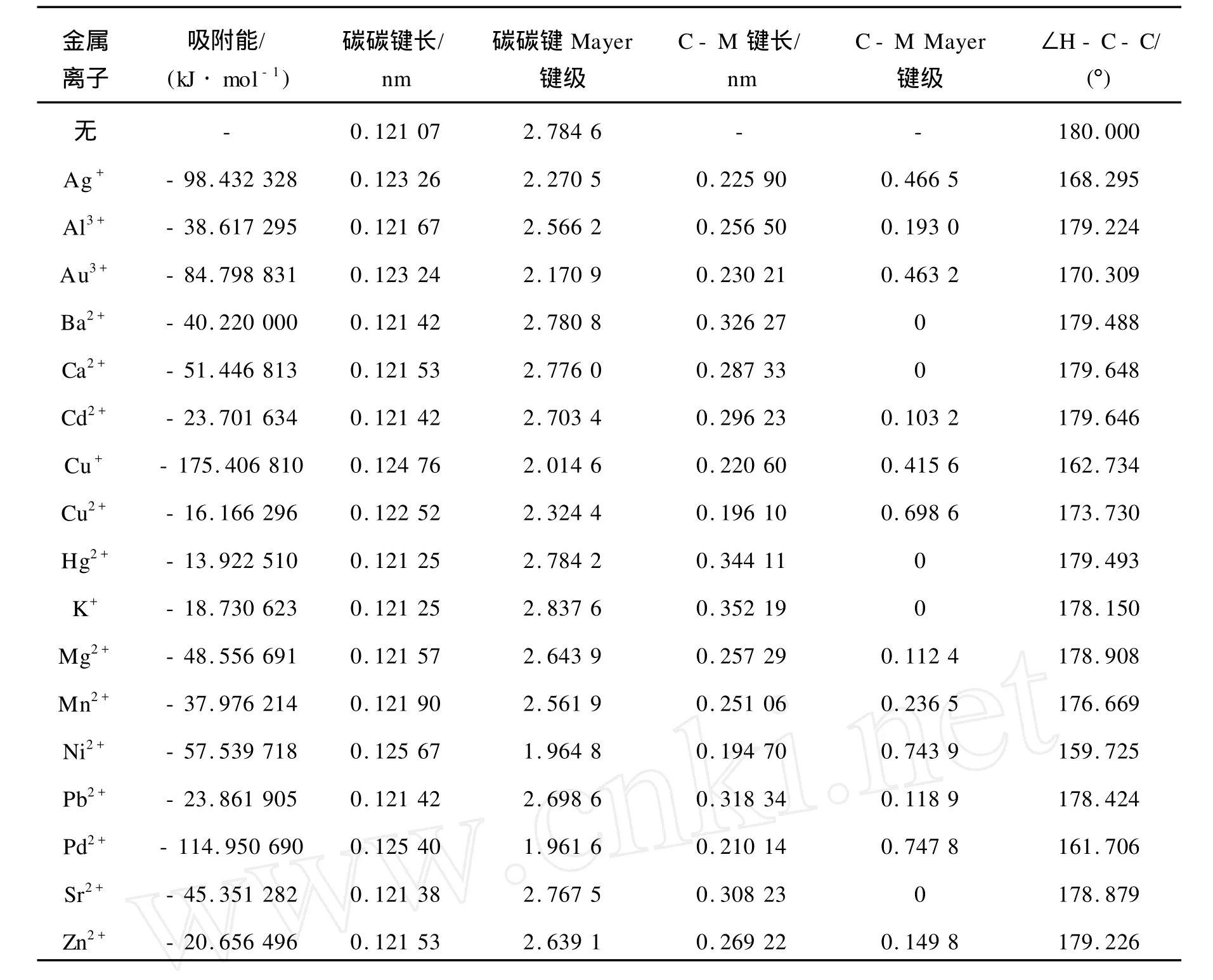
表1 金属离子与乙炔吸附前后的吸附能和结构参数Table 1 Adsorption energiesand optim ized structure parameters of adsorption of acetylene on variousmetal ions
图2为乙炔碳碳叁键键长与金属Mayer价指数变化的关系。由图2可知,乙炔吸附前后,金属Mayer价指数变化越大,碳碳叁键越长。同时,在金属氯化物的作用下,乙炔的两个氢原子向远离金属氯化物的方向偏移,从而使乙炔的H-C-C有一个小角度的非直线平面结构,这样有利于碳碳叁键的π轨道电子与金属的价轨道作用。
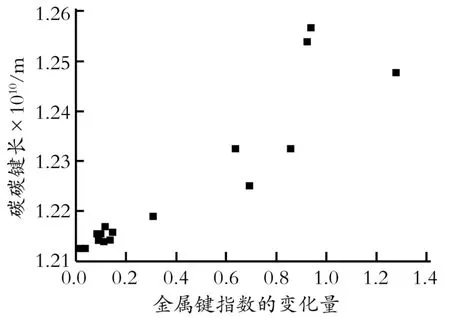
Fig.2 Dependence of the length of the CC triple bond of acetylene on the changes of Mayer total valence图2 乙炔碳碳叁键键长与金属Mayer价指数变化的关系
乙炔在各金属氯化物上的吸附络合作用放出吸附热,使得络合体系稳定。由表1可知,AgCl、 AuCl3、CuCl、NiCl2、PdCl2的吸附能比较高,乙炔与其发生的是化学吸附,形成比较稳定的化学键,同时由Mayer键级可知,乙炔吸附在这些金属氯化物上时,乙炔的碳碳叁键已经转变为了碳碳双键,金属离子与碳原子间的距离也达到了成键距离,由Mayer键级也可知金属离子与碳原子之间的键级均大于0.4,可以认为形成了离子键,其中,NiCl2和PdCl2与碳原子的键级大于0.7,形成更为稳定的化合价。
2.2 络合作用对乙炔的影响
当金属与乙炔发生作用时,金属的电子组态会发生变化,从而导致价电子的变化,同时也会影响到形成化学键时对成键电子吸引力的强弱。电负性越高的金属元素,对电子的吸引能力越强,既对乙炔的吸附越强。乙炔吸附前后金属M ayer价指数变化的大小可以反映出电子变化的多少,对Mayer价指数和键级的研究就可以预测化学键的断裂,进而可以预测反应进行的难易程度[13]。
图3为乙炔碳碳叁键外层电子布局数随金属M ayer价指数变化的规律。由图3可知,当金属Mayer价指数在吸附过程中变化不大时,碳碳叁键的电子布局数也基本保持不变或有升高,这说明由于金属离子的作用,使得乙炔内部出现了电子的转移,乙炔上氢原子的电子集中到了碳碳叁键上,使得碳原子的电子布局数升高,而金属离子的M ayer价指数没有明显的变化。当金属Mayer价指数改变较大时,M ayer价指数改变大于0.6的有AgCl、AuCl3、CuCl、CuCl2、NiCl2、PdCl2,吸附后的乙炔有较大的电子流失,碳原子上的电子流向了金属原子上,使得金属原子的价指数升高,与乙炔形成了稳定的化学吸附。
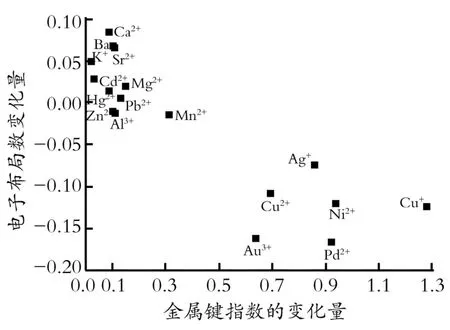
Fig.3 Dependence of Mulliken population of the CC triple bond of acetylene on the changes of Mayer total valence图3 乙炔碳碳叁键价层电子布局数与金属M ayer价指数变化的规律
2.3 不同金属对乙炔活化效率的预测
对于某一化学反应可能有很多种催化剂具有活性,哪种催化剂的活性最高与化学吸附强度有关[14]。乙炔与金属氯化物的反应生成中间体,其反应活性与乙炔在金属氯化物上的吸附特性有关,故分析它们的吸附络合可以达到预测反应的目的。
图4为乙炔碳原子的M ulliken电荷随金属Mayer价指数的变化规律。由图4可以看出,随着吸附后金属价指数的升高,乙炔碳原子所带的M ulliken电荷数也在增加,对碱土金属元素和大多数的二价过渡金属氯化物而言,乙炔碳原子M ulliken电荷数随M ayer价指数的变化基本呈线性,碳原子的M ulliken电荷数随金属价键指数的升高而升高。纯态的乙炔中碳原子的M ulliken电荷为-0.094,图中大多数的金属离子吸附乙炔后,乙炔碳原子的M ulliken电荷数下降,说明乙炔中的碳原子电子增加。但是结合图3可知,金属离子的电荷变化不大,所以乙炔碳原子上电子的富集是氢原子提供的。
图5为乙炔碳原子的M ulliken电荷随金属的M ulliken电荷的变化规律。由图5可知,乙炔吸附在金属氯化物上时,金属离子所带的负电荷越少,C -M键越强,乙炔受到的影响越大,碳碳叁键的活性也就越强。由图5也可以看出,金属氯化物与乙炔作用后,其吸附络合行为可分为3个区域:图中左上角几个金属的氯化物对乙炔的吸附作用较强,碳原子失去电子,电子转移到了金属上,并与金属原子形成了稳定的化学键,要打破这种稳定会比较困难,所以不宜继续发生反应;位于中间位置的金属离子形成的氯化物对乙炔的吸附作用适中,金属离子和碳原子之间有一定的电子转移,产生诱导效应,电荷比较集中于乙炔的碳碳叁键上,使乙炔分子得到了很好的活化;右下角三个金属的氯化物对乙炔发生吸附作用时,乙炔中碳原子所带的电荷较多,而金属离子上所带的电荷数降低,使得金属离子孤立出来,与氯原子的作用变小,如图1所示。而乙炔中碳原子的负电荷数却很高,这可能会改变催化反应历程,使得乙炔与金属氯化物的络合体容易与亲电性较强的氯化氢直接作用反应,而不经过氯乙烯金属氯化物的中间体。
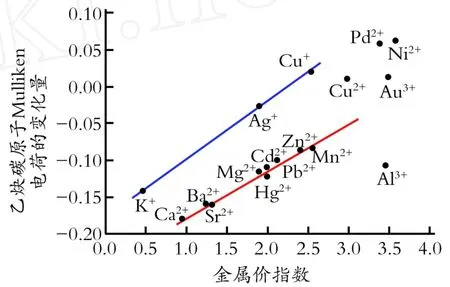
Fig.4 Dependence of the Mulliken charge of carbon atoms of acetylene on metal Mayer total valence图4 乙炔碳原子的M ulliken电荷随金属Mayer价指数的变化规律

Fig.5 Dependence of the Mulliken charge ofcarbon atoms of acetylene on the changes of metal Mulliken charge图5 乙炔碳原子的M ulliken电荷随金属的M ulliken电荷的变化规律
参考文献
[1] A rmentrout PB,Halle L F,Beauchamp J L.Periodic trends in transition metal-hydrogen,metal-carbon,and metaloxygen bond dissociation energies.Co rrelation w ith reactivity and electronic structure[J].J.Am.chem.soc.,1981,103 (21):6501-6502.
[2] Elkind J L,A rmentrout PB.Does ground state Fe+(iron(1+))reactw ith hydrogen[J].J.Am.chem.soc.,1986,108 (10):2765-2767.
[3] A ristov N,A rmentrout PB.Reaction mechanism s and thermochemistry of vanadium ionsw ith ethane,ethene and ethyne [J].J.Am.chem.soc.,1986,108(8):1806-1819.
[4] Georgiadis R,A rmentrout P B.Reactions of ground state Cr+w ith C2H6,C2H4,cyclo-C3H6,and cyclo-C2H4O: bond energies for CrCH+n(n=1-3)[J].International journal of mass spectrometry and ion p rocesses,1989,89(2-3): 227-247.
[5] Fisher E R,A rmentrout PB.Heat of formation of the hydroperoxyl radical HO2.A direct determination from guided ion beam studies of oxygen-methane[O2+(2.PI.g,v=0)+CH4]reaction[J].J.phys.chem.,1990,94(11):4396-4398.
[6] Ling Jiang,Qiang Xu.Observation of anomalous C—O bond weakening on discandium and activation p rocess to CO dissociation[J].J.Am.chem.soc.,2005,127(1):42-43.
[7] Walters R S,Schleyer P R,Corminboeuf C,et al.Structural trends in transition metal cation-acetylene comp lexes revealed through the C—H stretching fundamentals[J].J.Am.chem.soc.,2005,127(4):1100-1101.
[8] Walters R S,Pillai E D,Schleyer P R,et al.Vibrational spectroscopy and structures of Ni+(C2H2)n(n=1-4) comp lexes[J].J.Am.chem.soc.,2005,127(48):17030-17042.
[9] 杨亚丽,陆春海,黄娟,等.水分子在立方ZrO2(110)面吸附与解离的密度泛函理论研究[J].催化学报,2009,30(4): 328-334.
[10] M itchenko S A,Krasnyakova T V,M itchenko R S,et al.Acetylene catalytic hydrochlo rination over pow der catalyst p repared by p re-milling of K2PtCl4salt[J].Journal of molecular catalysis A:Chemical,2007,275(9):101-108.
[11] Conte M,Carley A F,Heirene C,et al.Hydrochlo rination of acetylene using a supported gold catalyst:A study of the reaction mechanism[J].Journal of catalysis,2007,250(9):231-239.
[12] 张旭之.乙烯衍生物工学[M].北京:化学工业出版社,1995.
[13] Mayer I.Bond o rder and valence indices:A personal account[J].J.comput.chem.,2007,28(1):204-221.
[14] 赵振国.吸附作用应用原理[M].北京:化学工业出版社,2005.
(Ed.:SGL,Z)

孙兆林,男,汉族,1962年6月生,山东泰安人,教授、博士生导师,现任中共辽宁省葫芦岛市委副书记、市长。曾任教育部化学与化工类专业教学指导委员会委员,辽宁省人民政府学位委员会第二、三、四届学科评议组成员,全国高协组织教材研究与编写委员会委员,《中国化工报》理事会副理事长,《石油知识》全国理事会理事、副主任委员,辽宁石油化工大学校长等职务。享受国务院政府特殊津贴,被选为“国家百千万人才工程”第一、二层次和辽宁省“百千万人才工程”百人层次,先后被授予“全国高等学校优秀骨干教师”、“辽宁省优秀专家”和“辽宁省劳动模范”等光荣称号。主要研究方向:新型催化材料,化学计量学与计算化学,分析化学,微波化学。
First-Princip les Studies of Acetylene Comp lexation With Various M etal Chlo rides
L IQiang1,JIANG Heng1,D ING Yong2,CHEN Yong-chang2,SUN Zhao-lin1*
(1.L iaoning Key Laboratory of Petrochem ical,L iaoning Shihua University,Fushun L iaoning 113001,P.R.China; 2.Chem istry and Chem ical Engineering,Lanzhou University,Lanzhou Gansu 730000,P.R.China)
1 June 2010;revised 7 July 2010;accepted 9 July 2010
Density functional theory(DFT)calculations were carried out fo r the op timization of the acetylene comp lexation w ith variousmetal chlo rides.Electronic structures were calculated and analyzed comp rehensively.The results show that ionic interactions between acetylene carbon-carbon trip le bond and the metal atom s has been found,w hich activate the acetylene. Dependence of geometry structure,adso rp tion energies and electronic changes of acetylene on the changes of Mayer total valence is also calculated.Activation p ropertiesof acetyleneover variousmetal ionsare discussed to p rovide theoretical basis for the research of non-mercury catalyst in the reaction of acetylene hydrochlo rination.
Acetylene;Metal chloride;Comp lexation;First-p rincip les
TQ032.41
A
10.3696/j.issn.1006-396X.2010.03.007
1006-396X(2010)03-0032-05
2010-06-01
李强(1984-),男,黑龙江虎林市,在读硕士。
辽宁省高等学校科技创新团队课题(2007T107)。
*通讯联系人。
*Co rresponding author.Tel.:+86-413-6860658;fax:+86-413-6860658;e-mail:lsong@lnpu.edu.cn
