Bi0.5Ca0.5Mn1-xCoxO3体系中的电荷有序和相分离
2010-09-08王强
王强
(陕西教育学院数理工程系,西安710061)
(2009年11月5日收到;2010年3月7日收到修改稿)
Bi0.5Ca0.5Mn1-xCoxO3体系中的电荷有序和相分离
王强†
(陕西教育学院数理工程系,西安710061)
(2009年11月5日收到;2010年3月7日收到修改稿)
利用固相反应法制备了Bi0.5Ca0.5Mn1-xCoxO3(0≤x≤0.12)系列多晶样品.研究了Co掺杂对Bi0.5Ca0.5MnO3电荷有序的影响.结果表明,Co掺杂导致电荷有序相逐渐融化、铁磁相互作用的增强;当x≥0.08时,电荷有序转变峰完全消失,但残留的反铁磁电荷有序相总是存在.相分离即反铁磁电荷有序和铁磁相共存对体系在低温下的性质具有重要作用.此外,发现Co比Cr更有效地破坏Bi0.5Ca0.5MnO3的电荷有序,这不同于稀土锰氧化物.
钙钛矿锰氧化物,电荷有序,团簇玻璃,相分离
PACC:7470V,7530H,7550L,6480
1. 引言
钙钛矿混价锰氧化物T1-xDxMnO3(T为La,Pr,Bi等,D为Ca,Sr,Ba)是一种典型的强关联材料体系,该体系中的电子、自旋、轨道与晶格之间存在强烈的相互作用,并因此导致了一系列奇特的物理现象[1],如超大磁电阻效应、绝缘体—金属转变、电荷有序和相分离等.这些物理现象的发现极大地丰富了凝聚态物理的内涵,同时锰氧化物在磁传感器、磁随机存储器、自旋晶体管等自旋电子学领域具有很好的应用前景,从而备受人们的关注.
尽管人们对该体系的结构、磁性、电输运性质等的变化规律已经有了比较深入的研究,但是锰氧化物体系还有很多深层次的物理问题有待解决,如电荷有序相产生的物理机理问题.电荷有序是实空间中Mn3+和Mn4+离子的周期排列,电荷有序相变通常与磁化强度的峰值、电阻的突变、比热和声速等的急剧变化相联系[2—4].以往对电荷有序的研究主要集中于诸如La1-xCaxMnO3等稀土锰氧化物,对铋基锰氧化物Bi1-x(Ca,Sr)xMnO3的研究相对较少.与稀土锰氧化物比较,铋基锰氧化物电荷有序转变温度普遍很高,最高达600K[5].相应地,破坏这些电荷有序相所需要的磁场也很高.例如,Bi0.5Ca0.5MnO3的电荷有序相变温度为TCO=325 K,破坏其电荷有序相需要高达560 kOe(1 Oe=79.5775 A/m)的磁场[6].在稀土锰氧化物中,单电子带宽W和电荷有序温度TCO可由A位平均离子半径调控,A位平均离子半径越大,单电子带宽W越大,这样会促进eg电子的传导,从而降低电荷有序相变温度.这一简单的规律对于稀土锰氧化物普遍适用,而对铋基锰氧化物却不成立.这是由于Bi3+存在高度极化的6s2孤对电子,它能引起的晶格畸变,这样就抑制了eg电子的传导,促成反铁磁电荷有序相的形成[7].
实验研究发现,在Mn位用其他过渡金属离子(如Cr,Co,Ni等)掺杂是破坏电荷有序的有效方法[8,9].Co离子与Mn离子半径相近,而且Co离子具有三种不同的价态和自旋态.具有不同价态和自旋态的Co离子对Mn的替代,可能会带来很多奇特的物理现象,能为探索电荷有序态稳定化的机理积累更多的实验数据.本文研究了Bi0.5Ca0.5Mn1-xCoxO3(0≤x≤0.12)系列样品的磁化强度、电阻与温度的关系等.结果表明,Co掺杂能抑制电荷有序、增强铁磁相互作用.与以前报道的结果相比较,发现Co比Cr更有效压制Bi0.5Ca0.5MnO3的电荷有序转变,这与稀土锰氧化物不同.
2. 实验方法
系列多晶样品Bi0.5Ca0.5Mn1-xCoxO3(x=0,0.015,0.03,0.05,0.08,0.12,0.16)采用常规的固相反应法制备.按化学计量配比将高纯度的Bi2O3,CaCO3,MnO2,Co2O3充分研磨使其混合均匀.然后在800℃,900℃温度下煅烧24 h;煅烧后经过研磨并压片,最后在1000℃下煅烧12 h,随炉冷却至室温,即得所需样品.
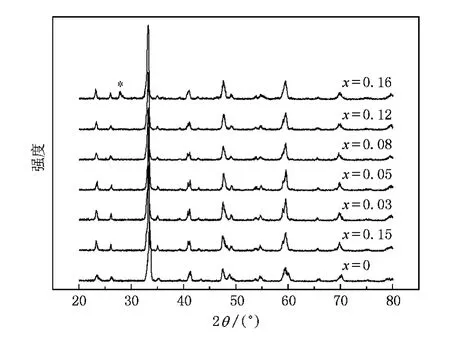
图1 室温下Bi0.5Ca0.5Mn1-xCoxO3(0≤x≤0.16)的X射线衍射谱(*表示杂相)
用MXP18AHF型转靶X射线衍射仪在室温下测得样品的X射线衍射谱,结果如图1所示.当x≤0.12时,能得到单相正交结构的样品;而当x=0.16时,X射线衍射谱出现了额外杂峰(用﹡标出),这与文献[10]报道的结果相同.使用MPMS-5型超导量子磁强计(SQUID)测量样品的磁性.电阻采用标准的四引线法在Quantum Design公司的PPMS-9系统上测量.
3. 结果与讨论
3.1. 磁性质
在1 kOe磁场下升温时测量了样品的MFC和MZFC磁化曲线(图2).从图2可以看到,对于x=0的样品,磁化曲线在TCO=325 K出现一个尖峰.Bao等人[11]根据中子散射的结果将该峰解释为电荷有序相变.TCO以上的温区,磁化率满足Curie-Weiss定律χ=C/(T-θP),其中θP=147.25 K>0,说明存在短程铁磁(FM)关联.一般认为这一短程关联来自eg电子在Mn3+和Mn4+之间跳跃产生的双交换相互作用[12].在电荷有序相载流子跳跃不再发生,因而FM关联被抑制,并为反铁磁(AFM)关联替代.当温度降至TN=130K,磁化曲线再次出现峰值,它是长程AFM序的标志[12].

图2 在1 kOe磁场下样品Bi0.5Ca0.5Mn1-xCoxO3(0≤x≤0.12)的直流磁化强度MFC和MZFC随温度T的变化关系
随着Co掺杂量的增加,电荷有序转变峰变弱并向低温移动,说明Co掺杂抑制了电荷有序相变;对于x≥0.08的样品,电荷有序转变峰完全消失.Co掺杂对AFM相变的影响更为突出,当x=0.015时,AFM相变仅表现为一个平台.当x≥0.03时,通过磁化曲线已经无法探测到AFM相变相关的结构,表明Co掺杂破坏了长程AFM序.在锰氧化物中较强的AFM相互作用有利于电荷有序相的形成,同样反过来逐渐变弱的AFM相互作用能导致了电荷有序转变峰的消失.
我们再来看低温磁性随Co掺杂量的演变.对于母体样品,当温度降至40K附近,MFC和MZFC曲线均陡然上升,这可能是出现短程FM关联的结果. MZFC曲线在Tf(Tf~35 K)存在一个显著尖峰,通常称Tf为自旋冻结温度.在Tf温度以下,磁矩开始集体冻结.MFC和MZFC曲线在Tf处出现分岔现象,这是自旋玻璃(spin glass,SG)或团簇玻璃(cluster glass,CG)的特征[13].对典型的自旋玻璃,在Tf以下MFC曲线不随温度变化[14],而这里在Tf以下MFC还在继续增加,并不显示一个平台,所以这里的玻璃转变其实是团簇玻璃转变,且发生FM性关联的温度与玻璃转变温度相近.另外,MFC和MZFC曲线在Tf≤T≤TN范围相重合,说明在TN以下为纯的AFM相变.这与以前的研究结果一致[15].随着掺杂量的增加,MFC和MZFC曲线在较高的温度出现分岔现象,分岔温度Td表明FM团簇中FM有序的开始.这时候的AFM不再是单纯的,其中存在FM性团簇[16].低温下MFC曲线的磁化强度值随掺杂量的增加而增大,说明FM相互作用的增强.对于样品x=0.12,已经有FM转变的迹象,而且这个转变温度(TC=180K)比玻璃转变温度高许多,具有再入型自旋玻璃(Reentrant spin glass)行为[17].
为了更深入地理解Co掺杂对磁性的影响,我们将MFC数据画成1/χ-T关系曲线,并根据Curie-Weiss定律(χ=C/(T-θP))在T

图3 Curie-Weiss温度θP值与Co含量x的关系
3.2. 电输运性质
实验研究[10,19]已经表明Mn位掺杂的Bi0.5Ca0.5MnO3的电输运性质能用可变程跃迁(VRH)模型ρ =ρ0exp(T0/T)1/4进行拟合.于是我们做了所有样品的lnρ-T-1/4曲线,如图4所示.对于x=0,电阻在325 K突然迅速增加,标志着电荷有序相变的发生,这与磁测量的结果相对应.在电荷有序相变温度以下,电阻随温度的降低迅速增大,这是由于电荷有序相变抑制了载流子的传导.在AFM相变温度以下,电阻的突变并不十分明显.在温度低于80K,由于电阻值超出了仪器测量范围,没有获得该温区的电阻.
随着掺杂量的增加,电阻的突变变得越来越模糊并向低温移动,表明Co掺杂抑制了电荷有序相变,这与磁测量的结果相一致.样品在低温下的lnρ-T-1/4曲线都能用VRH模型拟合,表明这些样品是典型的两相甚至多相混合的不均匀态[20],这与样品的相分离行为即AFM电荷有序相和FM相共存相一致.与此相反,在较高的温度lnρ-T-1/4曲线随掺杂量的增加逐渐偏离VRH模型.
磁性测量结果已表明,Bi0.5Ca0.5MnO3在高温区存在短程FM关联,这种FM关联通常用无规则分布并波动的局域化的极化子来描述[21].这样载流子在这些局域化的FM关联之间无序跃迁,从而lnρ-T-1/4曲线能用VRH模型拟合.随着Co掺杂量的增加,载流子能获得足够的热能穿越势垒并跃迁到最近邻位,因此lnρ-T-1/4曲线偏离VRH模型.相反,在较低的温度,载流子不能穿越势垒,从而lnρ-T-1/4曲线在低温区仍然满足VRH模型.

图4 零场下实验样品的lnρ-T-1/4曲线,虚线是可变程跃迁模型的拟合结果
3.3. 铋基锰氧化物与稀土锰氧化物Mn位掺杂效应的比较
关于Mn位元素掺杂对稀土锰氧化物Re0.5Ca0.5MnO3(Re:La,Pr,Nd等)电荷有序的影响人们已经做了比较深入的研究.对于Re0.5Ca0.5MnO3,在所有Mn位掺杂元素中Cr能最有效地破坏电荷有序;TCO随Cr掺杂量的增加线性降低,每1%的Cr能使TCO降低约8 K;TCO越高,完全破坏电荷有序相需要的Cr含量就越大[10].按照这样的规律,Xiong等[10]估计需要约20%的Cr才能完全抑制Bi0.5Ca0.5MnO3的电荷有序.最近的研究[19]表明12%的Cr完全破坏了Bi0.5Ca0.5MnO3的电荷有序转变.本文的数据则显示8%的Co完全抑制Bi0.5Ca0.5MnO3的电荷有序转变,说明Co比Cr能更有效压制铋基锰氧化物的电荷有序转变,这与稀土锰氧化物不同.其次,对于Re0.5Ca0.5MnO3,随Cr,Co掺杂量的逐渐增加能迅速诱导出很强的铁磁性,并出现金属—绝缘体转变.例如,Barnabé等[22]报道了Cr和Co最优掺杂时Pr0.5Ca0.5Mn1-xMxO3(M=Cr,Co)在5 K温度下每个Mn位的磁矩分别为3μB,2.5μB,接近自旋完全平行排列的预期值3.5μB.而对于铋基锰氧化物,在5 K温度下Bi0.5Ca0.5Mn0.88M0.12O3(M=Cr,Co)(x= 0.12时磁矩最大)的每个Mn位的磁矩仅为0.58μB,0.05μB,远远小于3.5μB.由此可见,在Bi0.5Ca0.5Mn1-xMxO3体系中,反铁磁电荷有序相并没有被完全融化,反铁磁性仍然很强,载流子是局域化的,而不像稀土锰氧化物Mn位掺杂能诱导FM金属基态.
4. 结论
通过磁性和电输运性质的测量,系统地研究了Co掺杂对Bi0.5Ca0.5MnO3电荷有序的影响.磁性测量发现,Co掺杂导致了电荷有序相的逐渐融化,电荷有序转变峰变弱并向低温移动,当x≥0.08时,电荷有序转变峰完全消失.此外,随着Co掺杂量的增加,反铁磁相互作用减弱,铁磁相互作用增强,体系在低温下始终出现团簇态玻璃相.体系的电输运行为可采用VRH模型拟合,证实了体系的相分离行为,即由反铁磁电荷有序相和铁磁相组成的不均匀态.相分离对体系在低温下的性质具有重要作用.通过铋基锰氧化物与稀土锰氧化物Mn位掺杂效应的比较发现,Co比Cr能更有效地破坏Bi0.5Ca0.5MnO3的电荷有序,这违背了稀土锰氧化物的一般规律.
[1]Salamon M B,Jaime M 2001 Rev.Mod.Phys.73 583
[2]Li X J,Wang Q 2009 Acta Phys.Sin.58 6482(in Chinese)[李晓娟、王强2009物理学报58 6482]
[3]Ramirez A P,Schiffer P,Cheong S W,Chen C H,Bao W,Palstra T T M,Gammel P L,Bishop D J,Zegarski B 1996 Phys.Rev.Lett.76 3188
[4]Gao H P,Li B,Yu Y,Ruan K Q,Wu B M 2004 Acta Phys. Sin.53 3853(in Chinese)[高惠平、李波、余勇、阮可青、吴柏枚2004物理学报53 3853]
[6]Kirste A,Goiran M,Respaud M,Vanaken J,Broto J M,Rakoto H,von Ortenberg M,Frontera C,García-Muoz J L 2003 Phys.Rev.B 67 134413
[8]Machida A,Moritomo Y,Ohoyama K,Katsufuji T,Nakamura A 2002 Phys.Rev.B 65 064435
[9]Guo H Y,Liu N,Cai Z R,Zhang Y H 2006 Acta Phys.Sin.55 865(in Chinese)[郭焕银、刘宁、蔡之让、张裕恒2006物理学报55 865]
[10]Xiong C M,Sun J R,Li R W,Zhang S Y,Zhao T Y,Shen B G 2004 J.Appl.Phys.95 1336
[11]Bao W,Axe J D,Chen C H,Cheong S W 1997 Phys.Rev. Lett.78 543
[12]Woo H,Tyson T A,Croft M,Cheong S W,Woicik J C 2001 Phys.Rev.B 63 134412
[13]Zheng G H,Sun Y P,Zhu X B,Song W H 2006 Solid State Commun.137 326
[14]Binder K,Young A P 1986 Rev.Mod.Phys.58 801
[15]Tzankov D,Kovacheva D,Krezhov K,Puz'niak R,Wis'niewski A,Sváb E,Mikhov M 2005 J.Phys.:Condens.Matter.17 4319
[16]Zhao J J,Xing R,Lu Y,Haosibayar,Zhao M Y,Jin X,Zheng L,Ning W,Sun Y,Cheng Z H 2008 Chin.Phys.B 17 2021
[17]Dho J,Kim W S,Hur N H 2002 Phys.Rev.Lett.89 027202
[18]Yu Q Y,Zhang J C,Jia R R,Jing C,Cao S X 2008 Acta Phys. Sin.57 453(in Chinese)[於乾英、张金仓、贾蓉蓉、敬超、曹世勋2008物理学报57 453]
[19]Li X J,Wang Q 2009 Physica B 404 3703
[20]Volkov N,Petrakovskii G,Patrin K,Sablina K,Eremin E,Vasiliev V,Vasiliev A,Molokeev M,Bni P,Clementyev E 2006 Phys.Rev.B 73 104401
[21]Lu W J,Sun Y P,Zhu X B,Song W H,Du J J 2006 Solid State Commun.138 200
[22]Barnabé A,Maignan A,Hervieu M,Damay F,Martin C,Raveau B 1997 Appl.Phys.Lett.71 3907
PACC:7470V,7530H,7550L,6480
†E-mail:qwangxj@yahoo.com.cn
Charge order and phase separation in Bi0.5Ca0.5Mn1-xCoxO3system
Wang Qiang†
(Department of Mathematical and Physical Engineering,Shaanxi Institute of Education,Xi’an710061,China)
(Received 5 November 2009;revised manuscript received 7 March 2010)
Polycrystalline Bi0.5Ca0.5Mn1-xCoxO3(0≤x≤0.12)samples are synthesized by using the solid-state reaction.The effects of Co-doping on charge order of Bi0.5Ca0.5MnO3are studied.The results show that Co doping leads to the melting of the charge order and the enhancement of the ferromagnetic correlation.For x≥0.08,the charge order transition is completely suppressed;however,there are still remaining antiferromagnetic domains inside the system.The phase separation or the coexistence of the charge order and ferromagnetic phase induced by Co-doping plays an important role in the low temperature properties for the system.Moreover,unlike in the case of rare-earth manganites,Co is more efficient to suppress charge order of Bi0.5Ca0.5MnO3than Cr.
perovskite manganites,charge ordering,cluster glasses,phase separation
book=554,ebook=554
†E-mail:qwangxj@yahoo.com.cn
