HPLC法测定盐酸左旋多巴甲酯的含量研究
2010-07-27潘俏凤
潘俏凤
(广州医学院第三附属医院,广东广州 510150)
盐酸左旋多巴甲酯是左旋多巴衍生化的产物,为抗震颤麻痹类神经性药物左旋多巴的前体药物,具有易溶于水、易进入血-脑屏障,易制成各种口服剂型和注射剂,吸收快,生物利用度高等优点[1],但有关盐酸左旋多巴甲酯含量的测定方法目前报道少见,本文采用高效液相色谱法对盐酸左旋多巴甲酯进行了含量测定的研究,建立含量测定方法,旨在为盐酸左旋多巴甲酯的质量控制提供理论依据。
1 仪器与试药
1.1 仪器
Agilent 1100型高效液相色谱仪,配有Agilent Eelipse XDB-C18(4.6 mm×250 mm,5 μm)色谱柱、emstation 化学工作站(Agilent);FA 1004型电子天平(上海精科天平仪器厂);KS-6000超声波清洗机(宁波科声仪器厂);ZK-82A型真空干燥箱(上海市实验仪器总厂);TDL-40B台式离心机(上海安亭科学仪器厂);电热恒温水浴锅(上海博迅实业有限公司医疗设备厂)。
1.2 试药
盐酸左旋多巴甲酯标准品(批号:20091145,中国药品生物制品检定所);盐酸左旋多巴甲酯样品(批号:2009014、2009015、2009016,自制);甲醇(色谱醇),三氟乙酸(分析纯),磷酸(分析纯),盐酸(分析纯),无水乙醇(分析醇)。实验用水为双重蒸馏水。所有试剂均经0.45 μm的微孔滤膜过滤并超声脱气。
2 方法与结果
2.1 对照品溶液的制备
准确称取干燥至恒重盐酸左旋多巴甲酯对照品25 mg,以流动相作溶剂,配成浓度为0.25 mg/ml的对照品溶液,放入冰箱贮存,备用。
2.2 供试品溶液的制备
取盐酸左旋多巴甲酯供试品25 mg,精密称定,置100 ml量瓶中,加流动相溶解并稀释至刻度,摇匀,用0.45 μm的微孔滤膜过滤,即得供试品溶液。
2.3 色谱条件与系统适用性试验
采用 Agilent Eelipse XDB-C18(4.6 mm×250 mm,5 μm)色谱柱;流动相:甲醇-0.05 mol/L磷酸二氢钾(用磷酸调节pH=3.5)=20∶80;流速:1.0 ml/min;检测波长:280 nm;柱温:20℃;进样量:20 μl。取上述制备的对照品溶液、供试品溶液各20 μl,分别注入液相色谱仪,记录色谱图,盐酸左旋多巴甲酯保留时间约为 4.5 min,理论板数大于3000,在此色谱条件下,盐酸左旋多巴甲酯色谱峰达到基线分离。结果见图1。
2.4 线性关系的考察
精密取标准溶液 1、2、4、6、8 ml分别置 10 ml量瓶中,以流动相稀释至刻度,摇匀,配制成不同浓度的溶液,分别吸取上述溶液各20 μl,分别进样测定,以峰面积为纵坐标(Y),质量浓度(μg/ml)为横坐标(X)进行线性回归,得线性方程:Y=12072X-83.53(r=0.9999),盐酸左旋多巴甲酯进样量在25.65~256.50 μg范围内与峰面积呈良好线性关系。
2.5 精密度试验
按“2.1”项下方法配制对照品溶液,重复进样5次,结果盐酸左旋多巴甲酯峰面积的RSD=0.99%(n=5),表明仪器精密度良好。
2.6 稳定性试验

图1 HPLC色谱图(A.对照品;B.供试品)Fig.1 HPLC chromatography(A.Reference substance;B.Sample)
精密吸取标准品溶液适量,在室温下放置,于0、2、4、6、8、12、24 h 分别进样,并测定峰面积。 结果 RSD=0.69%(n=5),表明样品溶液在24 h内稳定性良好。
2.7 重复性试验
取盐酸左旋多巴甲酯样品(批号:2009014)适量,按“2.2”项下方法配制样品溶液,按上述色谱条件测定,记录色谱图,计算盐酸左旋多巴甲酯含量。结果RSD=0.872%,表明本法重现性良好。
2.8 加样回收率试验
精密称取已知含量的样品6份,每份约10 mg,分别加入盐酸左旋多巴甲酯对照品适量,按“2.2”项下方法制备溶液,按上述色谱条件测定,平均回收率为99.02%,RSD=1.34%(n=6)。表明本法回收率较好,结果见表1。
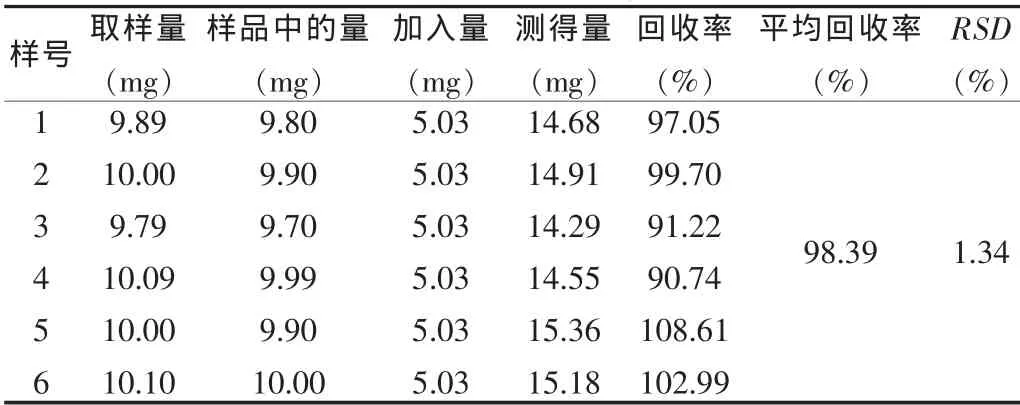
表1 回收率试验结果(n=6)Tab.1 Results of recovery test(n=6)
2.9 样品含量测定
取不同批号的盐酸左旋多巴甲酯样品(批号:2009014、2009015、2009016),各 3 份,按“2.2”项下方法制备样品溶液,按上述设定的色谱条件测定样品中盐酸左旋多巴甲酯的含量,结果见表2。

表2 样品含量测定结果(%,n=3)Tab.2 Content determination results of samples(%,n=3)
3 讨论
左旋多巴是人体内合成去甲肾上腺素及多巴胺等的前体物,能透过血-脑屏障进入脑内,经多巴脱羧酶转化成多巴胺,增加大脑纹状体中多巴胺含量,从而成为目前最有效的帕金森病(PD)的治疗药物[2],但左旋多巴在体内难以进入人体的血-脑屏障,因此对其进行结构修饰,对其进行衍生化成盐酸左旋多巴甲酯[3]。
目前,有关药物分析的方法较多,但由于HPLC和其他分离分析方法相比,具有柱效高、灵敏度高、分离速度快、适用范围广、重现性好、操作方便等优点,很快在药物分析中被广泛的应用,成为药物质量控制的重要手段[4-9]。实践表明,HPLC是一种非常有效和普遍适用的药物成分分析方法。本文在实验过程中较详细地考查了色谱分离检测参数,包括流动相的组成比例、检测波长、柱温和流速等对盐酸左旋多巴甲酯分离测定的影响。盐酸左旋多巴甲酯紫外检测在280 nm处有最大吸收且吸收值较稳定,不受盐酸浓度的影响,故本研究以280 nm波长用于其含量的分析。在流动相的选择中,笔者对流动相中甲醇与缓冲溶液的比例及缓冲溶液的pH值进行了优化,两者均对保留时间有较大影响,pH值对峰形也有较大影响,在pH值为3.5时得到最佳的峰形。通过本实验,笔者得出了盐酸左旋多巴甲酯的最佳色谱分离条件,并对样品分析的回收率和精密度进行了测定,建立了盐酸左旋多巴甲酯含量测定的HPLC测定方法,该法操作重复性良好,准确度较高,因此,该法能用于盐酸左旋多巴甲酯的质量控制。
[1]董宝平.左旋多巴与帕金森氏病[J].化学教育,2004,12(6):8-10.
[2]龙启才,袁进,赵树进.左旋多巴治疗帕金森病致症状波动的药动学机制[J].中国药房,2001,12(6):374-375.
[3]王建,谢林,王大为,等.HPLC法测定人血浆中左旋多巴和卡比多巴浓度及其药代动力学研究[J].中国药科大学学报,2004,35(3):239-243.
[4]宋晓凯,吴立军.天然药物化学[M].北京:化学工业出版社,2004:15.
[5]周岐勋,李健和,徐幸民.高效液相色谱法测定盐酸甲氯芬酯分散片含量及有关物质[J].中国当代医药,2009,16(4):73-76.
[6]关强,王云龙,杜遵义.高效液相色谱法测定延胡索药材中延胡索乙素的含量[J].中国当代医药,2009,16(4):78-79.
[7]刘丽,赵玉江,柳森国.HPLC法测定刺五加注射液中总异嗪皮啶的含量[J].中国现代医生,2007,45(9):52-53.
[8]刘慧明.高效液相色谱技术及其在药物分析中的应用[J].中国现代医生,2007,45(20):110.
[9]王娟.HPLC法测定匹卡米隆分散片的有关物质[J].中国现代医生,2007,45(8):68,77.
