克隆启动子方法的比较及应用
2010-07-09郝迪陈李淼李文滨
郝迪 ,赵 琳,陈李淼,李文滨
(东北农业大学大豆生物学教育部重点实验室,大豆研究所,东北农业大学农学院,哈尔滨 150030)
随着基因工程的发展,常常需要构建一种能高水平表达异源蛋白质的表达载体。启动子对外源基因的表达水平影响很大,是RNA聚合酶能够识别并与其结合、从而起始基因转录的一段DNA序列,是基因工程表达载体的重要元件。基因启动子的克隆,对研究植物基因表达调控和构建表达载体至关重要。文章就近几年来克隆植物基因启动子的各种方法做一综述,为启动子分离技术的应用提供理论参考。
1 启动子克隆的几种方法
1.1 PCR技术克隆启动子
对于序列已知的启动子,即根据发表的基因序列,设计引物,用PCR技术克隆基因的启动子。由于PCR法简便快捷,近年来被较多应用于克隆基因启动子。
Wang等根据GenBank中拟南芥基因组序列中ats1A(核酮糖-1,5-二磷酸羧化酶小亚基)基因的前部疑似启动子区域设计引物,以拟南芥总DNA为模板,PCR扩增出ats1A的基因启动子区片段[1]。苏宁等根据已报道的水稻叶绿体16S rRNA基因前部疑似启动子区域设计引物,以水稻叶绿体DNA为模板,PCR扩增出16S rRNA基因5'启动子区的片段。同源比较结果表明,所克隆的片段与水稻叶绿体16S rRNA启动子序列具有100%的同源性。可见,该方法简便、快捷、操作简单,但不能克隆到全新的启动子[2]。
1.2 I-PCR技术
I-PCR又称反向PCR(Inverse-PCR),是1988年由Triglia最早提出的一种基于PCR的改进的染色体步行方法。I-PCR的试验程序包括,基因组DNA经酶切后用T4DNA连接酶进行自连接,产生环状DNA片段;以环化产物为底物,用根据已知片段设计的反向引物进行PCR扩增,从而得到含有未知片段的扩增产物,见图1[3]。
Forester等用该方法克隆了豌豆种子脂肪加氧酶基因启动子,约800 bp片段[4]。韩志勇等以IPCR技术为基础克隆了转基因水稻的外源基因旁侧序列[5]。
I-PCR法快速、高效、稳定,操作相对简单,花费少,PCR引物设计比较方便。
1.3 P-PCR技术
P-PCR(Panhandle PCR)是由Jones等提出的利用末端反向重复序列与已知序列互补配对形成环状单链模板,有效增强了引物与模板结合的特异性。反应需要3个根据已知序列设计的引物,3个引物在已知序列内呈线性排列,其中第3个引物可作为接头使用,可与已知序列互补配对形成锅柄状单链模板(见图 2)[6]。
黄君健等成功地应用P-PCR技术从正常的人外周血单核细胞基因组DNA中扩增端粒催化亚基hTERT基因5'端上游旁侧序列,获得了hTERT基因翻译启始位点上游2 090 bp的基因组DNA序列[7]。

图1 I-PCR原理Fig.1 Sketch map of I-PCR
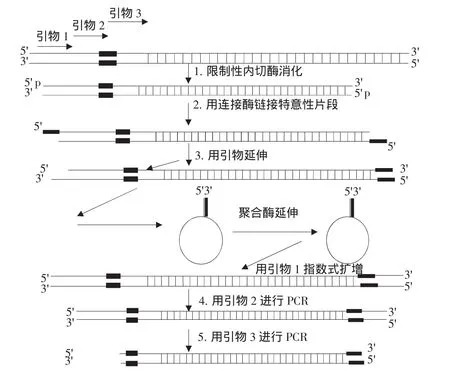
图2 锅柄PCR原理Fig.2 Sketch map of P-PCR
P-PCR是目前能够扩增距已知序列最远的未知DNA序列的方法,有很高的特异性。
1.4 锚定PCR技术
锚定 PCR(Anchored PCR)是由Huang等提出的扩增已知序列侧翼未知片段的方法。反应中需要2个根据已知序列设计的引物,2个根据载体序列设计的引物,然后分别利用一条已知序列特异性引物和载体上非特异性引物进行PCR扩增。其操作一般包括3轮PCR扩增,第1轮采用不对称PCR的方法,在扩增时使用两种引物浓度不同,一般高浓度引物与低浓度引物比为(50~100)∶1,在最初10~15个循环的产物主要是双链DNA,待低浓度引物耗尽后,继续由高浓度引物介导产生大量单链DNA。第2轮中,把第1轮PCR产物稀释后做模板,引物为相同浓度的特异性引物和非特异性引物,这一轮可得到双链DNA。第3轮PCR目的主要是检测所得PCR产物是否为预期产物,结果见图3[8]。
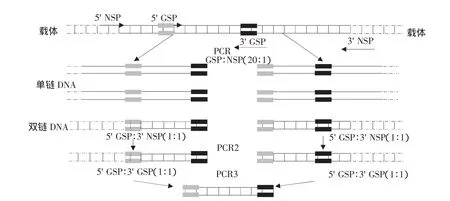
图3 锚定PCR的流程Fig.3 Schematic diagram of anchored PCR method
范云等用锚定PCR克隆了胡萝卜AFP(抗冻蛋白)基因上游序列[9]。
此方法需要知道启动子相邻序列,且要构建基因组文库。
1.5 探针载体筛选启动子
利用启动子缺失的报告基因构建启动子探针质粒,通过报告基因的表达来克隆有启动功能的DNA片段。在该技术中关键是要构建一种启动子探针载体。探针型载体是一种有效、经济、快速分离基因启动子的工具型载体,包含2个基本部分:转化单元和检测单元。其中,转化单元含复制起点和抗生素抗性基因,用于选择被转化的细胞;检测单元则包括1个已失去转录功能且易于检测的遗传标记基因以及克隆位点。
利用启动子探针载体筛选启动子的过程为,先选用1种适当的限制性核酸内切酶消化切割染色体DNA,然后将切割产生的DNA限制片段群体与无启动子的探针质粒载体重组,并按照设计的要求使克隆的片段恰好插在紧邻报告基因的上游位置;随后再把重组混合物转化给寄主细胞,构建质粒载体基因文库,并检测报告基因的表达活性。
最早由Rachael等在大肠杆菌中以四环素抗性基因作为报告基因构建了启动子探针质粒pBRH3B,并克隆了一些原核和真核启动子片段。其后Donna等以氯霉素抗性基因作为报告基因,Fodor等以大肠杆菌LacZ为报告基因,构建了酵母启动子探针质粒并克隆了一些启动子片段。张晓宁等用启动子探针载体pECE7克隆了盐藻耐盐基因的启动子[10-13]。
该方法不需要知道具体基因的序列,可随机筛选启动子,避免了引物设计,能获得大量的启动子片段。
1.6 利用载体或接头的染色体步行技术克隆基因启动子
该方法首先要提取基因组DNA,用限制性内切酶消化,产生出适合于克隆的DNA片段,然后在体外将这些DNA片段同适当的λ噬菌体载体连接成重组体分子,并转化到大肠杆菌的受体细胞中,构建基因组文库。将构建好的基因组文库铺板,转移到尼龙膜上,用部分已知序列的片段或特异的基因片段做探针进行杂交,筛选出具有目的启动子的克隆,测序分析即可获得该启动子。
王新国等利用衔接头的方法[14],设计了位于单链DNA两端互补的颠倒末端重复序列,增加了反应的特异性,在胡萝卜II型转化酶基因启动子的克隆方面取得了新的进展。
这种方法具有便于操作、试验线路简单的优点,但是特异性较差,产物需进一步杂交验证。
1.7 YADE法
Prashar等在扩增cDNA 3'端时采用“Y”形接头,以减少接头引物的单引物扩增[15]。
其原理是接头引物处于“Y”接头的2个分叉单链上,序列与接头一样,只有与特异引物引导合成了接头的互补序列后,接头引物才能退火参与扩增(见图 4)。
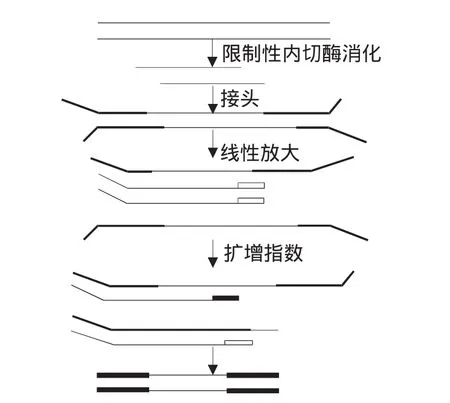
图4 YADE法的流程Fig.4 Schemtic diagram of YADE
方卫国等尝试将YADE法引入到昆虫病原真菌的分子生物学研究[16]。在已克隆的类球孢白僵菌类枯草杆菌蛋白酶基因CDEP-1的基础上,利用YADE法,克隆到该基因的启动子CDEPP。过程是先酶切球孢白僵菌基因组DNA,然后与“Y”形接头相连,取连接产物做模板,先以基因特异引物1做线性扩增,再以线性扩增产物为模板,以接头引物和基因特异引物2做指数扩增,只有当线性扩增时合成了含有接头引物的互补单链,接头引物才能与其发生退火,参与指数扩增,从而有效防止了接头引物的单引物扩增。最后得PCR产物,进行序列分析确定为CDEP-1的上游启动子序列。
在应用YADE法时,内切酶的选择至关重要。好的内切酶产生适合PCR扩增的片段,太大太小都不行。为了得到合适的内切酶,需要从众多的内切酶中筛选。研究表明,不同的物种有自己合适的内切酶。YADE法延伸的起始片段可以是基因组DNA片段,也可以是cDNA片段,在延伸cDNA片段时,设计的引物需要避开内含子和外显子的边界,在内含子的位置未知的情况下,可考虑多合成1~2条特异引物,以提高扩增未知片段的机率。该方法假阳性低、效率高,理论上能扩出所有目的片段。
1.8 接头PCR技术
接头PCR是继I-PCR方法之后的又一用来扩增基因组中已知序列两侧未知序列的方法。当基因序列已知,要克隆此基因启动子时即可使用此方法,其原理如下:首先,用几种限制性内切酶酶切DNA,然后将酶切后的DNA与体外合成的接头在适当的条件下连接,取适量连接产物直接作为PCR模板,其中一条引物为接头特异引物,其序列能与接头序列互补;另一条引物为基因特异引物,其序列与已知序列互补,该引物3'端朝向要扩增的序列区。最后将PCR产物克隆并测序。
在这种方法中,有一个特殊的接头,该接头由一条长链和短链组成,短链接头的3'端有一个NH2的存在,它能阻断短接头链的酶促延伸,而且只有当一条远端的基因特异引物相对长链接头方向延伸一条DNA链后,才能产生引物结合位点。如果产生了两末端都含有双链接头序列的PCR产物(非特异性PCR产物),由于反向重复序列的存在,在紧接着的每步变性过程中DNA链的末端会形成发夹结构。这些结构比引物与模板的杂交链更稳定,因此会抑制指数式扩增。然后,当一个远端的基因特异性引物通过接头延伸一条DNA链,延伸产物将只有一端会有接头序列,这样就不能形成发夹结构,PCR扩增能正常进行。
王新国等利用这种特殊的接头设计了位于单链DNA两端互补的颠倒末端重复序列,一条长链接头序列和一条短链接头序列,增加了反应的特异性,在胡萝卜Ⅱ型转化酶基因启动子的克隆方面取得了新的进展[14]。根据接头PCR的原理,TaKaRa公司推出了LA-PCR体外克隆试剂盒(DDR015)。用来克隆基因侧翼未知序列。Miao用试剂盒克隆了小麦淀粉合成酶基因gbss的启动子序列。Li等也用LA-PCR试剂盒的方法,首次分离了长度为1 216 bp的山葡萄几丁质酶基因VCH3上游启动子序列[17-18]。
1.9 TAlL-PCR技术
热不对称交错PCR(Thermal Asymmetric Interlaced PCR,简称TAIL-PCR)是一种用来分离与已知序列邻近的未知DNA序列的分子生物学技术。该技术由Liu和Whitter首先研究并报道。在分子生物学研究领域中,利用该技术分离出的DNA序列可以用于图位克隆、遗传图谱绘制的探针,也可以直接测序[19-20]。
TAIL-PCR技术的基本原理是利用目标序列旁的已知序列设计3个嵌套的特异性引物(specialprime,简称sp1,sp2,sp3,约20 bp),用它们分别和1个具有低Tm值的短的(14 bp)随机简并引物(Arbitrary Degenerate Prime,ADP)相组合,以基因组DNA作为模板,根据引物的长短和特异性的差异设计不对称的温度循环,通过分级反应来扩增特异引物。反应流程如图5所示。由一个特异性引物和一个简并引物组合构成的PCR反应叫做“半特异性PCR”。这种反应会产生3种不同类型的产物:①由特异性引物和简并引物扩增出的产物;②由同一特异性引物扩增出的产物;③由同一简并引物扩增出的产物。在TAIL-PCR反应中,后两种非目标产物可以通过以嵌套的特异性引物进行的后续反应来消除。
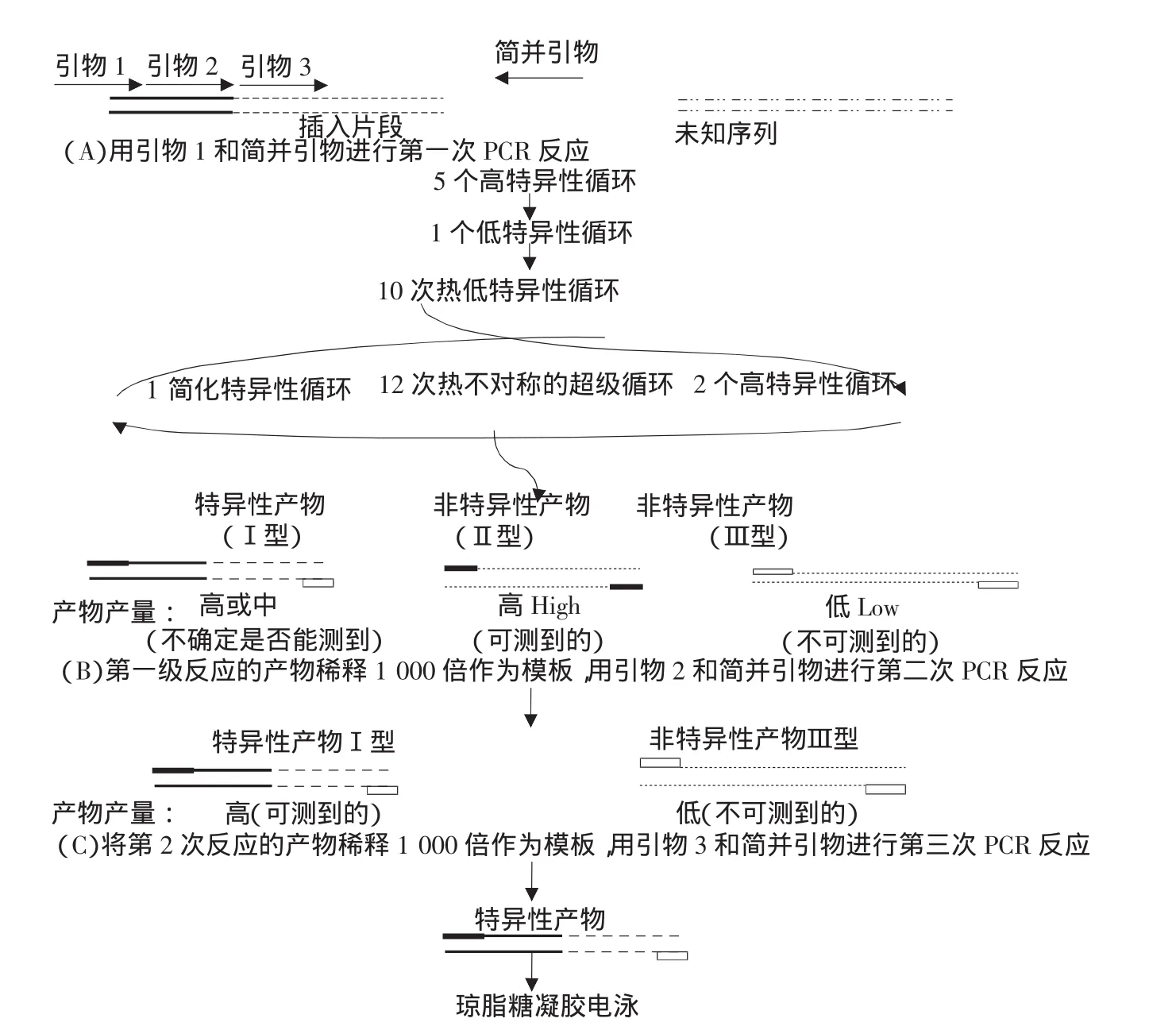
图5 TAIL-PCR反应流程Fig.5 Protocol of TAIL-PCR
TAIL-PCR反应的条件在不同的试验中稍有变化。在表1所示的条件下对P1和YAC克隆插入末端序列的分析中获得了满意的结果。这种反应条件亦是一个规范的试验设置,可以满足大多数试验的需要。反应体系中,特异性引物的浓度与普通的PCR相同,简并引物的浓度要高,一般为2.5~5 μmol·L-1,以满足引物的结合效率。(简并引物AD1:TGWGNAGWANCASAGA;AD2:CAWCGIC NGAIASGAA;AD3:TCSTICGNACITWGGA;AD4:NTCGASTWTSGWGTT;AD5:NGTCGASWGANAW GAA;AD6:WGTGNAGWANCANAGA;AD7:AG WGNAGWA NCAWAGG;D=G,A,T;M=A,C;N=A,G,C,T;R=A,G;S=G,C W=A,T)。
目前已成功地从P1、YAC和BAC克隆中分离获得插入末端的DNA序列,如表1的T-DNA侧翼序列。此外,经过改良的TAIL-PCR技术能够快速克隆Pal及Pgi基因的启动子序列和野油菜黄单胞菌群体感应信号基因。
TAIL-PCR技术能够快速地分离到目标序列,对基因克隆研究具有重要的意义。TAIL-PCR技术有以下优点:①简单。只要设计好引物,即可以用基因组DNA作模板直接筛选到目标序列,节省了PCR反应前后的许多费时、费力的操作程序。②特异性高。用短的简并引物和长的特异性嵌套引物相组合,通过不对称的温度循环和分级反应,使反应体系有利于特异引物的扩增,最终的扩增产物中目的片段占绝对优势,反应产物可以直接用做探针标记和测序模板。③高效灵敏。使用任何一个AD引物,在60%~80%的反应中能够产生特异性产物。运用不同的AD引物就能够有效地扩增到目标片段。④快速。整个TAIL-PCR反应循环能够在1 d内完成,可以快速地获得目标片段。⑤不涉及连接反应。反应产物准确可靠,重复性好。
2 启动子克隆方法比较
以上介绍的几种方法基本代表了现有的启动子克隆方法,它们分别具有不同的特点和适用范围,见表2。

表1 TAIL-PCR反应分离P1和YAC克隆的插入末端序列Table 1 Cycling conditions used for TAIL-PCR of P1and YAC clone
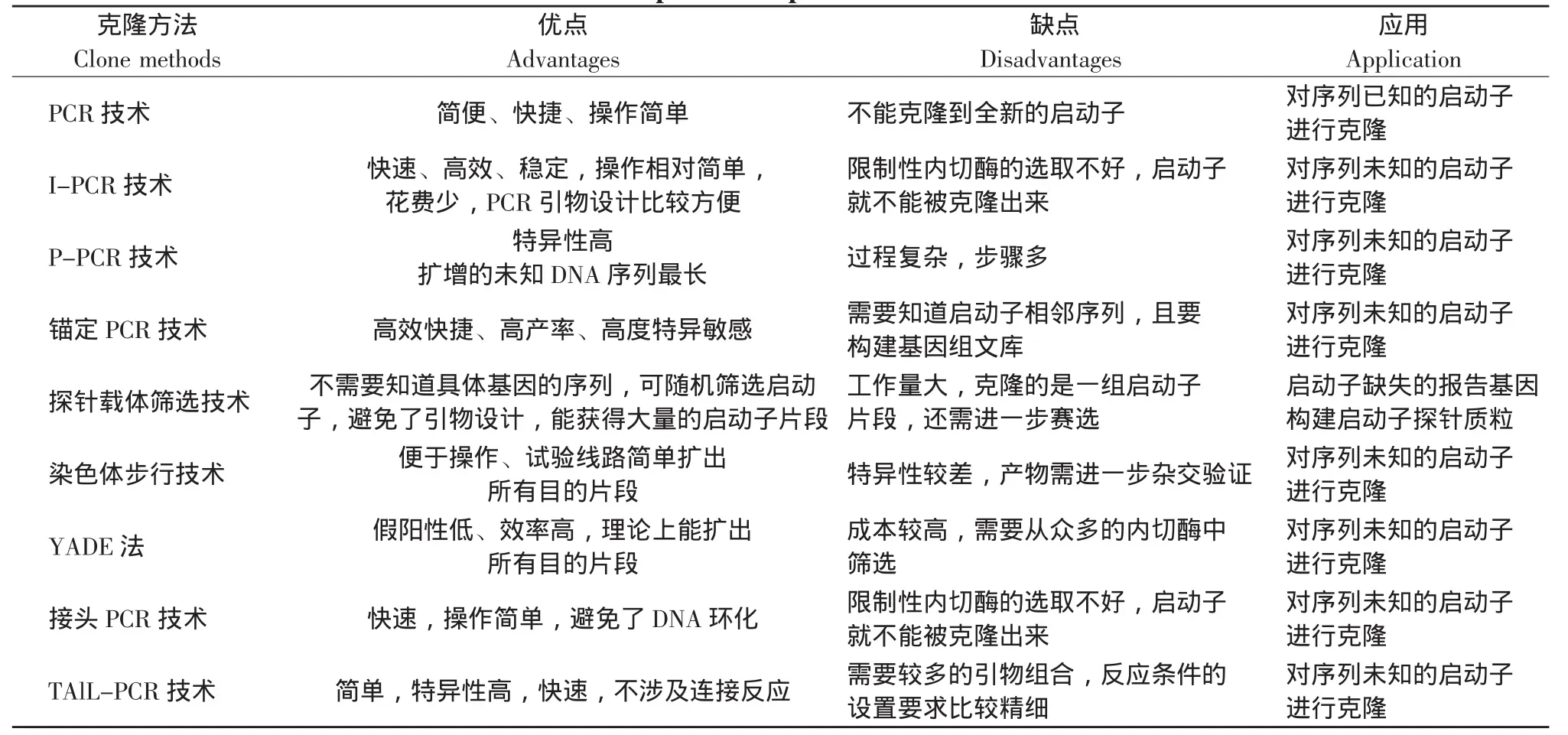
表2 启动子克隆方法比较Table 2 Comparison of promoter clone methods
3 展望
目前,启动子克隆的方法种类繁多,进展迅速。启动子克隆方法绝大多数是建立在PCR的基础上的染色体步行技术,因而利用启动子克隆的方法不仅可以克隆到基因的启动子,同时还可以用于克隆cDNA的全长。另外,随着基因工程的发展,出现了越来越多的转基因动植物,启动子克隆的方法也可以应用在转基因生物的研究中,如转基因水稻的T-DNA区,通过启动子克隆方法的PCR步行技术,能成功地克隆到T-DNA区的旁侧序列,有助于分析突变体中突变区序列。
随着PCR技术的不断进步,一定会有更多、更好的克隆启动子的方法产生,而它们最后也将会是克隆基因全长或突变体(特别是转座子插入突变体)中目的基因的简便、高效的新方法。
[1]Wang L J,Fan S H,Guo A G.Cloning and functioanalysis of ats1A gene promoter from Arabidopsis thaliana[J].Acta Botanica Boreali-Occidentali Sinica,2004,4(10):1856-1860.
[2]苏宁,孙萌,李轶女,等.水稻叶绿体16S启动子克隆改造、载体构建及转化研究[J].植物学通报,2003,20(3):295-301.
[3]TrigliaT,PetersoMG,KempDJ.AProcedurefor in vitro amplification of DNA segments that lie outside the boundaries of known sequences[J].Nuclic Acids Res,1988,16:8186-8186.
[4]ForesterC,ArthurE,CrespiS,etal.Isolationofapea(Pisumsativum)seed lipoxygenase promoter by inverse polymerase chain reaction and characterization of its expression in transgenic tobacco[J].Plant Mol Biol,1994,26(1):235-248.
[5]韩志勇,王新其,沈革志,等.反向PCR克隆转基因水稻的外源基因旁侧序列[J].上海农业学报,2001,17(2):27-32.
[6]Jones D.Panhandle PCR[J].PCR Meth Appl,1995,4:S195.
[7]黄君健,李杰之,林坚,等.人端粒酶催化亚基hTERT基因启动子的克隆[J].生物技术通讯,1999,10(3):36-39.
[8]Huang S H,Jong A Y,Yang W,et al.Amplification of gene ends from gene library by PCR with single-sided specificity[J].Methods Mol Biol,1993,15:357-363.
[9]范云,刘兵,王宏斌,等.胡萝卜抗冻蛋白基因的克隆及其在E.coli中的表达[J].中山大学学报,2002(3):26-29.
[10]Rachael L N,Robert W W,Raymond L R.Eukaryotic DNA fragments which act as promoters for a plasmid gene[J].Nature,1979,277:324-325.
[11]DonnaMW,ElizabethJD,PaulSL.Cloningrestrictionfragmentsthat promoteexpression of a gene in Bacillus subtilis[J].J Bacterol,1981,146:1162-1165.
[12]FordorI,KranikovaOV,BeretsE,etal.Cloningstructureandfeatures of a Saccharomyces cerevisiae DNAfragment causing the expression of reportergenes[J].Mol Biol,1990,24:1411-1418.
[13]张晓宁.盐藻耐盐相关基因的克隆、功能分析与高效启动子的分离鉴定[D].上海:复旦大学,2002:53-67.
[14]王新国,张国华.用衔接头PCR克隆新的胡萝卜Ⅱ型转化酶基因启动子[J].中国生物学与分子生物学报,2001,17(1):61-65.
[15]Prarshar Y,Weissman S M.the cloning of halotoler ant correlative gene,functionanalyse[J].ProcNatlAcadSciUSA,1996,93:659-663.
[16]方卫国.用YADE法克隆球孢白僵菌类枯草杆菌蛋白酶基因CDEP-1的启动子及启动子序列分析[J].菌物系统,2003,22(2):252-258.
[17]Miao H M.Cloning and characterization of the promoters of key starch synthesis enzyme genes in wheat[J].2004,85:57-68.
[18]Li H Y,Qi J,Shu H R,et al.Isolation and characterization of a chitinase gene VCH3promoter from grapevine[J].Plant Physiol Mol Biol,2005,31(5):485-491.
[19]Liu Y G,Robert F W.Thermal asymmetric interlaced PCR:automatable amplification and sequencing of insert end fragment from P1and YAC clones for chromosome walking[J].Genomics,1995,25:674-681.
[20]Liu Y G.Huang Ning Efficient amplification of insert end sequences from bacterial artificial chromosome clones by thermal asymmetric interlaced PCR[J].Plant Molecular Biology Reporter,1998,16(2):175-181.
