集成数据选择特征基因
2010-03-24王海鹏
王海鹏,杨 昆
(杭州电子科技大学计算机学院,浙江杭州310018)
0 引 言
利用基因芯片数据对疾病进行分类诊断是生物医学中重要的应用领域[1]。目前已有的基因选择方法大致可以分为3类:过滤法,缠绕法和嵌入式法[1]。集成分析来自相同研究问题的不同数据集是分析基因表达数据的一个可行途径。本文提出了一种新的集成数据选择特征基因的方法(Gene Selection by Multiple Data Integration,GSMDI),针对多来源数据中的每一个,首先计算每个基因在这一数据上的差异表达统计量,然后用每个基因的差异表达统计量来代替这一原始数据进行后面的分析,从而尽可能地消除数据本身带有的特异性。最后利用多来源的数据提取特征,在不同的单一来源的数据上进行训练和测试,训练分类器的数据和测试数据是同一来源的,不同来源的数据仅仅用于特征的提取。提出的基因选择方法有效地避免了原有的数据集成方式的不足。
1 基因选择方法
Ap×n1和Bp×n2是针对相同的科学问题而由不同的研究产生的两个微阵列数据,它们共同包含p个相同基因。对于这两个不同来源的数据集A、B,用基因选择方法(基于T统计量的方法[2])分别应用于单个数据集计算,得到基因对应的统计量xA,yB(p维列向量)。对于来自这两个数据集上的某个公共的基因g,若基因g在两个数据集上的表达一致,则在理想状态下应该有xA=yB,也就是说点(xA,yB)在二维直角坐标系中是位于y=x上的。在如图1所示的二维直角坐标系中,令x轴代表基因在其中一个数据集上的统计量,y轴代表该基因在另一个数据集上的统计量。那么找到可靠的特征基因的问题就转化为在(x,y)平面上找到离y=x距离最近的那些点。对于二维平面中的任意一个点O(x,y),满足如下的命题:
命题 对于不在中线上的坐标系中任意一点O(x,y),该点距离中线y=x的距离OC为|x-y|/2。
证明 如图1所示,过点O分别做平行于x轴和y轴的直线,与中线分别相交与点A、B。由几何知识得OA=OB=|x-y|,因为ΔOAB为直角三角形,所以斜边由三角形面积相等,得该点到中线的距离
对于二维直角坐标系中其他象限的任意点,该命题同样成立。

图1 几何表示
但是这种方法存在一定的问题:由于max{|x|,|y|}很小,距离原点越近的点,其距离中线的距离也越近,这样的基因用于分类没有多大的效果。所以,必须对|x-y进行修正,本文中的|x-y|/|x+y|就是其中的一种修正方式,由于|x+y|相对较大,在一定程度上避免了这种方法存在的问题。
以上是两个数据集的情况,对于n个数据集,基因选择方法——GSMDI(gene selection by multiple data integration)为:假设有n个基因表达数据集,首先用基因选择方法(基于T统计量的方法[2])应用于单个数据集计算,得到基因对应的统计量。再对不同数据集上的每个基因对应的统计量,两两组合,计算F=|x-y|/|x+y|。对每个基因对应的个F值取平均值,然后以平均值为准从小到大排列,依次取前50、100、150、200个基因作为选出的特征基因。最后采用KNN(K-最近邻)和SVM(支持向量机)分类器,在训练数据上训练,在测试数据上测试。
2 实验结果
2.1 数据集
本文实验的真实数据是来自4个关于乳腺癌的Affymetrix microarray数据,这4个数据是由不同的研究机构使用不同型号的Affymetrix寡核苷酸微阵列得到的。其中Westet al[3]的数据由HuGeneFL芯片产生,它包含两个类别共49个样本,其中25个为乳腺癌样本,24个为正常样本。Huang et al[4]的数据由HG-U95Av2芯片产生,包含两个类别共89个样本,其中74个为乳腺癌样本,15个为正常样本。Wang et al[5]的数据由HG-U133A芯片产生,包含两个类别共286个样本,其中209个为乳腺癌样本,77个为正常样本。Soiriou et al[6]的数据也是由HG-U133A芯片产生,包含两个类别共183个样本,其中149个为乳腺癌样本,34个为正常样本。由于产生数据的微阵列型号不同,用基于序列的探针匹配方法来匹配微阵列中使用的探针,找到公共的探针集。最终得到5 045个在3种微阵列芯片中同时出现的公共探针集,以这5 045个公共探针集在4个数据中的表达值为分析目标。
2.2 实验设置和数据分类
本文提出的算法用Matlab实现,通过分类实验来比较本文提出的方法GSMDI和作为对照的基因选择方法T-test。具体的实验环境是:Intel Core2 2.1 Ghz处理器,2G内存,操作系统是Windows XP。本文使用5-fold交叉验证实验来获得平均分类结果。实验结果部分所示的是50次5-fold交叉验证实验的平均结果。
对于实验中4个来源的数据集A、B、C、D,把它们分成A1+A2、B1+B2、C1+C2、D1+D2,定义*1为特征提取与训练分类器的数据,*2为测试数据(它不参与到特征提取与训练中)。对A、B、C、D4个数据集进行交叉验证实验,即每次把它们分成训练与测试集,对于一个具体的分割有A1+A2、B1+B2、C1+C2、D1+D2,在(A1,B1,C1,D1)上,用提出的方法来提取特征G,然后在特征G上分别以*为训练集,分类测试集*。作为对照实验,依次在单个数据集上用基于T统计量的方法来提取特征,然后以选出的特征为对象,在*上训练,分类*G2。
2.3 实验结果
实验结果从准确率、敏感性和特异性3个指标考察分类实验,相关的性能指标的定义如下:准确率确预测为正常的样本数,TN(true negative)是被正确预测为乳腺癌的样本数,FP(false positive)是被错误预测为正常的样本数,FN(false negative)是被错误预测为乳腺癌的样本数。

图2 在4个数据集上两种分类器的准确率
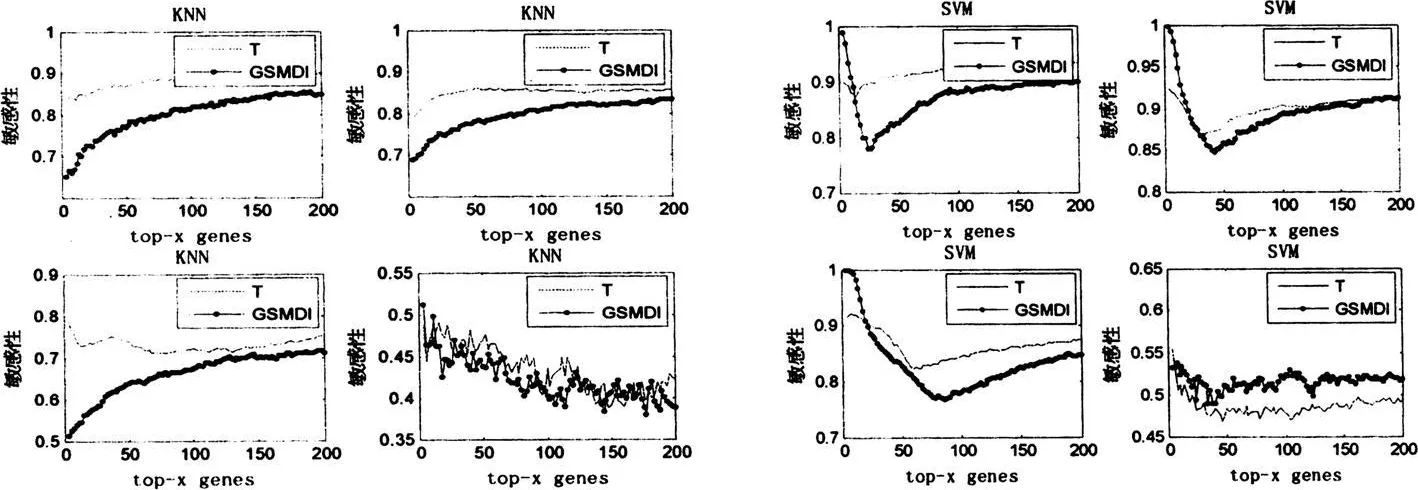
图3 在4个数据集上两种分类器的敏感性
图4 在4个数据集上两种分类器的特异性
实验结果见图2-图4,其中每幅图中包含左右两幅图,对应两种不同的分类器。对于每种不同的分类器各有4个小图,其中左上个子图表示Huang etal的数据,右上个子图表示Sotiriou etal的数据,左下个子图表示Wang et al的数据,右下个子图表示West etal的数据。
分类实验结果显示,尽管在某几个数据上对于单一的指标(比如图2的左上个子图,Accuracy指标,Huang et al的数据,SVM分类器)提出的方法不如对照的方法T-test。但是从总体上看,综合考虑所有的3个指标,所有的4个数据,2种不同的分类器,提出的方法是有优势的。
3 结束语
本文提出了一种新的集成数据选择特征基因的方法,针对多来源数据中的每一个,首先计算每个基因在这一数据上的差异表达统计量,然后用每个基因的差异表达统计量来代替这一原始数据进行后面的分析,最后利用多来源的数据提取特征,在不同的单一来源的数据上进行训练和测试。在4个真实的基因表达数据集上对提出的方法进行了测试,实验结果证明本文提出的方法所选出的特征应用于分类的效果更好。
[1] 周昉,何洁月.生物信息学中基因芯片的特征选择技术综述[J].计算机科学,2007,34(12):143-150.
[2] Varma S,Simon R.Iterative class discovery and feature selection using Minimal Spanning Trees[J].BMCBioinformatics,2004,(5):126.
[3] WestM,Blanchette C,Dressman H,etal.Predicting the clinical status of human breast cancerby using gene expression profiles[J].Proc Natl Acad Sci USA,2001,98(20):11 462-7.
[4] Huang E,Cheng SH,Dressman H,et al.Gene expression predictors of breast cancer outcomes[J].Lancet,2003,361(9369):1 590-6.
[5] Wang Y,Klijn JGM,Zhang Y,etal.Gene-expression profiles to predictdistantmetastasis of lymph-node-negative primary breast cancer[J].Lancet,2005,365(9460):671-9.
[6] Christos Sotiriou,PratyakshaWirapati,Sherene Loi,et al.Gene Expression Pro?ling in Breast Cancer:Understanding the Molecular Basis of Histologic Grade To Imp rove Prognosis[J].JNatl Cancer Inst,2006,4(98):262-72.
