球形硅基介孔分子筛的合成和表征
2010-01-31胡龙兴谌永峰徐佳佳
胡龙兴, 谌永峰, 徐佳佳
(上海大学环境与化学工程学院,上海 200444)
1992年,美国Mobil公司首先成功地利用烷基季铵盐阳离子表面活性剂为模板剂,合成出新型M41S系列氧化硅基有序介孔分子筛[1-2].该类分子筛具有较大的比表面和均一的孔道直径分布,孔径在 1.5~10 nm范围可调变,在催化、分离与吸附等方面有很广阔的应用前景[3-7].1998年,Zhao等[8]首次以聚环氧乙烯醚-聚环氧丙烯醚-聚环氧乙烯醚三嵌段共聚物 P123为模板剂,在强酸性条件下合成了SBA-15硅基介孔分子筛.该类介孔分子筛具有大孔径和厚孔壁等优点,因此具有很高的水热稳定性.随后,SBA-15受到越来越多研究者的关注,并迅速成为研究热点之一[9-15].
目前合成球形MCM-41和球形 SBA-15的方法较少[16-19].M.Grün等[16]通过简便方法合成球形MCM-41,但并未深入探讨球形MCM-41的合成机理.A.Katiyar等[18]通过水热方法合成球形 SBA-15,但并未对球形 SBA-15进行 TEM表征.根据文献[18]可以发现,合成的球形 SBA-15颗粒尺寸较大,无法直接通过 TEM观测其孔道结构.本研究在这两种方法的基础上合成出球形MCM-41和 SBA-15,并对合成的分子筛进行表征,通过研究MCM-41的合成过程来分析其合成机理,同时分析了湿磨法对球形MCM-41和球形SBA-15样品XRD,SEM和 TEM表征结果的影响.
1 实验部分
1.1 球形MCM-41介孔分子筛的合成
合成过程依据文献[16]的方法:先将0.75 g的CTAB溶解在 15 g的去离子水中,再将4.5 g的氨水(质量分数为 25%,0.066 mol/L)和23 mL的无水乙醇(EtOH)加入到表面活性剂溶液中,搅拌 15 min,随后加入 1.4 g的 TEOS,此时凝胶中各物质(TEOS,CTAB,NH3,H2O,EtOH)的摩尔比为 1∶0.25∶10∶123∶80.在室温下搅拌 2 h后,对样品溶液进行过滤、水洗、干燥处理,所得的白色粉末即为带有模板剂的MCM-41,最后对其进行煅烧,在空气气氛下,起始温度为室温,升温速率为 1 K/min,升温到823 K后恒温 6 h,随后是自然降温过程.
1.2 球形 SBA-15介孔分子筛的合成
合成过程参照文献[18]的方法,确定具体工艺条件,其典型合成过程如下:将 3 g P123溶于 60 mL (1.5 mol/dm3)的 HCl,然后将 0.6 g CTAB与 25 mL去离子水混合,并加入 20 mL乙醇.随后将10 mL TEOS逐滴加入到该表面活性剂溶液中,并将混合物在 45℃剧烈搅拌 (约 500 r·min-1)45 min.搅拌后,将混合液转移到反应釜中并于静态条件、75℃下放置 10 h,然后在 120℃下老化 48 h.最后进行煅烧,在 8 h内将温度缓慢升至 550℃,恒温煅烧 6 h后自然降温.
1.3 球形介孔分子筛的表征
采用日本的Max-2000型微区转靶 X衍射仪对介孔材料进行 XRD分析,Cu靶 (Kα1),Ni滤波λ =1.540 56,加速电压 40 kV,管电流 200 mA,确定其晶相及特征介孔结构.采用美国麦克仪器公司(Micromeritics)的 Tristar 3000型全自动氮吸附比表面积测试仪,通过气体吸附法测定样品的比表面积、孔体积和孔径分布.采用日本 JS M-6700F扫描电子显微镜和日本 JEM-2012F透射电子显微镜分别观测样品表面形貌和孔道结构.
2 结果与讨论
2.1 MCM-41的表征
2.1.1 XRD分析
XRD扫描角度为 1°~15°,其分析图谱如图 1所示.
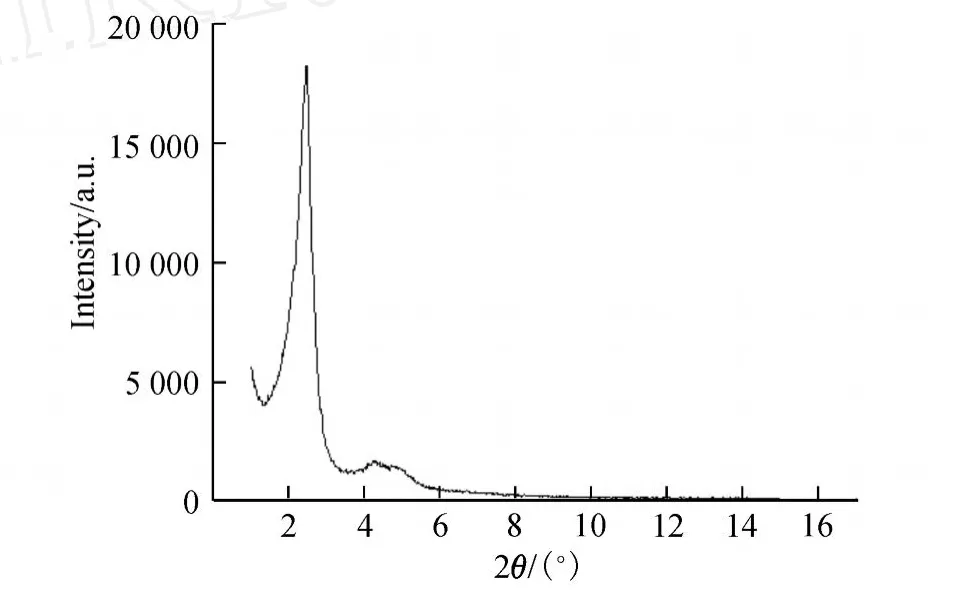
图1 合成的球形MCM-41的XRD图Fig.1 Low-angle XRD patterns of sphericalMCM-41
由图1可以看出,合成的MCM-41的图谱与典型MCM-41的图谱基本一致.XRD在小角区内出现了 3个 hk0衍射峰,这说明合成的MCM-41介孔材料具有较高的质量.XRD图谱中,最强峰对应着孔道六方排列的 (100)面,另外两个次峰分别对应的晶面是 (110)和 (200).图谱中主峰的峰强度也较高,这说明合成的MCM-41介孔材料内部具有较高的有序性.
2.1.2 N2吸附分析
MCM-41的N2吸附-脱附等温线如图 2所示.由图 2可以看出,等温线图属于Ⅳ类.在低压力下,吸附量几乎线性增加;在相对压力为0.22~0.32时,吸附量突然迅速增加,这是因为在材料孔道内有毛细管凝聚;在相对压力为0.33左右时,孔道内被填满;相对压力大于 0.33之后,吸附量基本不增加.
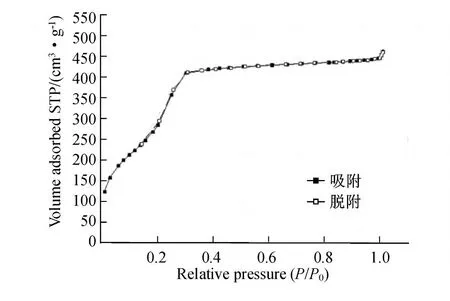
图2 合成的球形MCM-41的 N2吸附-脱附等温线Fig.2 N2adsorption-desorption isotherm s of spherical MCM-41
按一般吸附-脱附等温线的规律推断,该MCM-41应该具有 H1型迟滞环.但在本实验中未曾出现,原因是一般使用CTAB表面活性剂合成的MCM-41孔径不够大,只有孔径达一定值时,H1型迟滞环才会出现[20].
样品的比表面积为 1 107 m2·g-1,比孔容为0.69 cm3·g-1,孔径基本为 2~3 nm,平均孔径约为2.5 nm,这与文献[21]中的MCM-41 N2吸附-脱附实验结果相似.样品的平均孔径偏小,这是因为乙醇可以减少胶束头部基团之间的内部排斥作用并使胶束的直径变小.
2.1.3 SEM分析
MCM-41的 SEM分析如图 3所示.

图3 合成的球形MCM-41的SEM图Fig.3 SEM image of sphericalMCM-41
由图 3可以看出,本实验合成出了较好的球形介孔分子筛,其粒径尺寸基本分布在0.2~0.6μm.球形MCM-41的聚集情况比较严重,聚集体内的各单粒之间存在着一定程度的次生孔,这对于利用球形MCM-41进行吸附反应时会产生一定的影响,可能会增加吸附质的吸附量.
2.1.4 TEM分析
MCM-41的 TEM分析如图 4所示.

图4 合成的球形MCM-41的TEM图Fig.4 TEM image of sphericalMCM-41
由图 4可以看出,样品的中心区域均存在六方有序排列的结构,这与 XRD实验结果相符.另外,在球体的边缘部分,孔道并不是六方相排列,这是因为在测定过程中,这些孔道平行于电镜测定的方向,所以整体显示为中心向四周辐射的结构.该图与 B. Pauwels等[22]研究的结果相同.
2.1.5 球形MCM-41的合成机理
在合成过程中,首先在反应器中加入去离子水、CTAB、氨水和乙醇.在高的乙醇浓度下,通过搅拌,大量的乙醇分子吸附在 CTAB胶束的外表面,使得溶液与胶束中的乙醇分子的比例增大.此时,乙醇分子担任着共溶剂的角色,胶束分子不会大量聚集在一起,自动形成高界面曲率的球形胶束.此时氨水作为催化剂,催化水解随后加入的 TEOS.在氨催化剂的作用下,TEOS发生水解和聚合,形成硅基聚合物(见图 5).反应方程式如下:
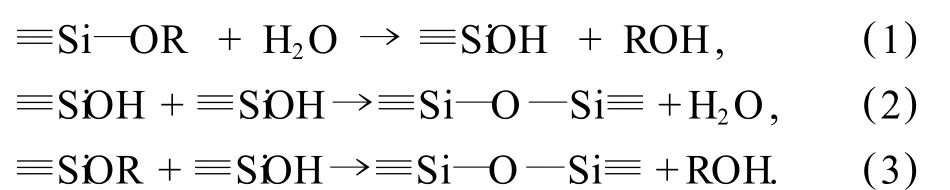
方程式(1)为水解反应,方程式(2)和(3)为缩合反应.
首先,经过水解反应,TEOS分子中的烷基被羟基基团取代.随后,该羟基基团中的一个氢离子去除,形成了硅酸盐阴离子.这时,带负电的硅酸盐阴离子就和带正电的 CTA+产生静电作用,而被吸附在胶束的表面.随后,由于吸附在胶束表面的硅酸盐阴离子之间的聚合作用,使得球形胶束分子向棒状胶束分子发展,同时各棒状胶束分子之间自组装形成一定的六方相结构.另外,硅酸盐阴离子之间的聚合作用还使得六方相结构的各胶束之间的空间进一步缩小,形成闭合的六方排列.当胶束长度进一步增大后,就会导致结构有序性下降,例如在MCM-41的TEM图中边缘部分的不规则的孔道结构.
在合成中,材料的形态取决于带负电的硅基胶束的聚合速率和介孔结构形成速率之间的平衡.在高浓度乙醇存在条件下,材料的形成过程受到表面张力的控制,只有通过形成球形颗粒才能最大限度地减小表面自由能,表面自由能减小的过程是自发过程[23].
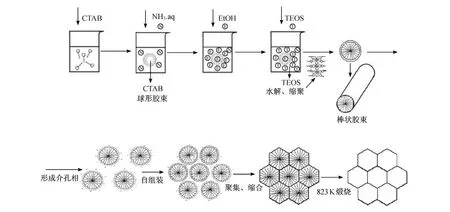
图5 MCM-41的合成机理Fig.5 Schematic view of the formation process ofMCM-41
2.2 SBA-15的表征
2.2.1 XRD分析
XRD的扫描角度为 0.5°~4.0°,其图谱如图 6所示.
由图 6可见,XRD图谱显示在 0.95°处有一很强的衍射峰,对应着 SBA-15的(100)面,峰稍宽,略微可看出对应着(110)的峰,但峰很宽.而对应着 SBA-15的(200)面的衍射峰则不可见,这表明在合成球形SBA-15过程中,一定程度地破坏了分子筛的有序结构.这可能是因为在合成过程中加入的 CTAB和乙醇与 P123胶束作用,降低了分子筛的有序性.
2.2.2 N2吸附分析
图7为样品的N2吸附-脱附等温线.由图7可见,合成的 SBA-15具有典型的Ⅳ型等温线和 H1型迟滞环,这表明样品具有规则的孔道结构.在低压力下,吸附量几乎线性增加,但增加较缓慢;在相对压力为 0.6~0.8时,吸附量突然迅速增加;在相对压力为0.8左右时,孔道达到饱和;在相对压力大于 0.8之后,吸附量基本不增加.制得的 S BA-15孔径约为7.4 nm,比表面积为 583 m2·g-1,比孔容为 1.27 cm3·g-1.
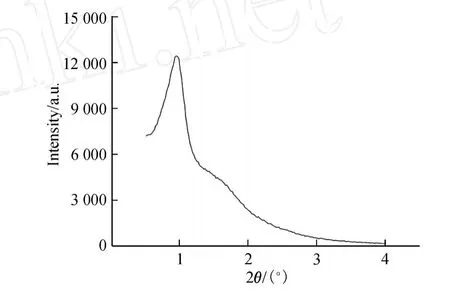
图6 合成的球形 SBA-15的 XRD图Fig.6 Low-angle XRD patterns of spherical SBA-15
2.2.3 SEM分析
图8为样品的 SEM图.由图 8可以明显看出,本实验合成的球形 SBA-15介孔分子筛相对于球形MCM-41来说,聚集程度明显较低,但其球形外貌相对稍差.SBA-15的粒径尺寸基本分布在 3~10μm,由于球形分子筛颗粒尺寸很大,导致直接通过 TEM方法无法得知分子筛的内部孔道结构.
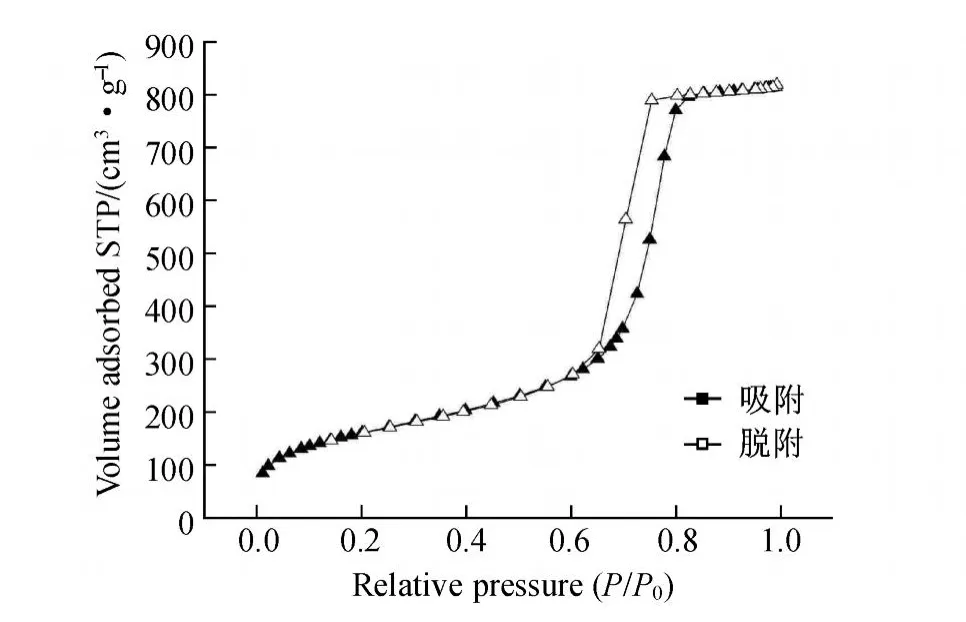
图7 合成的球形 SBA-15的 N2吸附-脱附等温线Fig.7 Nitrogen adsorption-desorption isotherm s of spherical SBA-15

图8 合成的球形 SBA-15的 SEM图Fig.8 SEM image of spherical SBA-15

图9 合成的球形 SBA-15的 TE M图Fig.9 TEM image of spherical SBA-15
2.2.4 TEM分析
SBA-15的 TEM图如图 9所示.由图 9可知,合成的球形分子筛尺寸非常大,达到微米级,而一般TEM方法只能观测尺寸在 500 nm以下的颗粒物,所以通过 TEM直接观测合成的球形分子筛不可行.我们尝试通过湿磨的方法将球形分子筛破碎,通过观察碎片来了解分子筛的内部孔道结构.但是,由于湿磨过程可能会改变原颗粒的内部结构,因此有必要考察湿磨后样品的表征结果.
2.3 湿磨对分子筛结构有序性的影响
2.3.1 对 XRD的影响
为了研究湿磨过程对样品内部孔道结构的影响,我们对湿磨后的球形 SBA-15进行了XRD表征,结果如图 10所示,其中曲线 a和 b分别为湿磨前和湿磨40 min后的 XRD图.
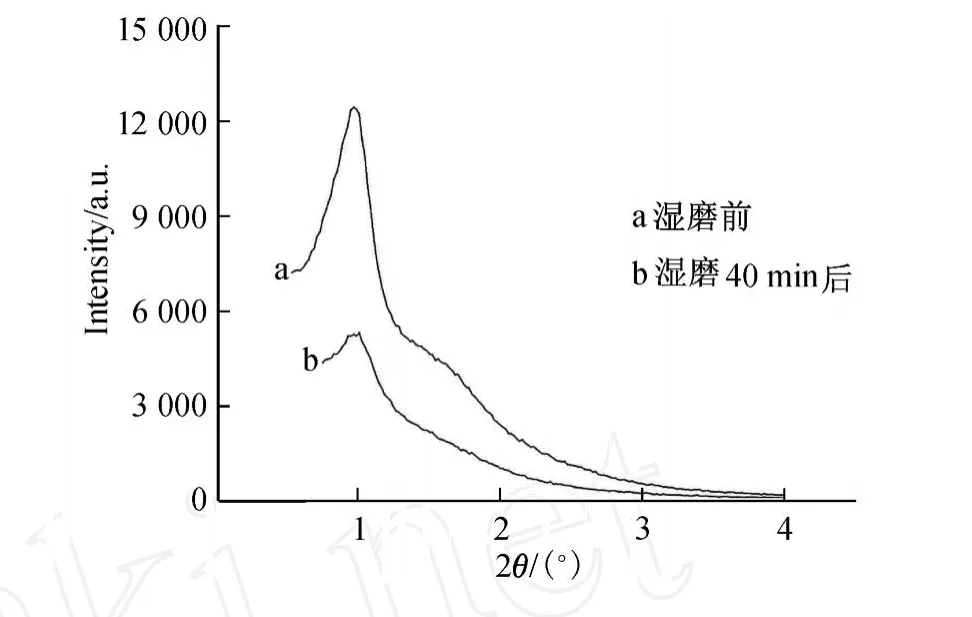
图10 湿磨过程对 SBA-15 XRD的影响Fig.10 Effect of wet grinding on XRD patterns of SBA-15
由图 10可见,样品经过湿磨后,其第一个特征峰有宽化的现象,且峰强度较湿磨前减弱,并且对应着 SBA-15的(110)和(200)面的衍射峰则完全不可见,这说明湿磨过程会影响样品内部结构的有序性,但并未破坏其二维六角的结构.
本实验未对湿磨后的MCM-41进行XRD表征,是因为由图 11可以看出,湿磨过程对MCM-41的球形外貌基本没有影响.
2.3.2 对 SEM的影响
样品的合成过程如 1.1节和 1.2节所述,研磨过程为湿磨法,湿磨 40 min,结果如图 11所示,其中图 11(a)和图 11(b)分别为MCM-41湿磨前和湿磨后的 SEM图,图 11(c)和图 11(d)分别为 SBA-15湿磨前和湿磨后的 SEM图.
由图11可以清楚看到,MCM-41因为颗粒尺寸较小,属于纳米级别,湿磨后球形颗粒基本没有被破坏,所以本实验所用的湿磨方法对其球形外貌基本没有影响.而 SBA-15因为颗粒尺寸较大,已达到微米级别,在本实验的湿磨方法下,其球形外貌已受到损害,颗粒已被完全破碎.由此可见,湿磨过程对不同粒径分子筛形貌的影响是不同的.
2.3.3 对 TEM的影响

图11 湿磨过程对MCM-41和 SBA-15球形外貌的影响Fig.11 Effect of wet gr inding on MCM-41 and SBA-15 spherical shapes
样品的合成过程如 1.2节所述,湿磨过程如2.3.2节所述,其结果如图 12所示,其中图 12(a)和图 12(b)为采用 Zhao等[8]的方法合成的麦穗状SBA-15湿磨前和湿磨后的 TEM图,图 12(c)为本实验合成的球形 SBA-15湿磨前的 SEM图,图 12 (d)和图 12(f)为本实验合成的球形 SBA-15湿磨后的TEM图.
图12(a)中表现出的长程有序性在图 12(b)中仍然可见,这说明湿磨过程对分子筛的孔道结构基本没有影响,也从另一方面证明了 2.3.1节中 SBA-15的 XRD表征的结果,即湿磨过程不会破坏 SBA-15的二维六角结构.而由图 12(c)~图 12(f)可以看出,经过湿磨后,TEM观测结果大多为图12(e)和图 12(f)的形式,球形 SBA-15的长程有序性的显示较少.这可能是因为球形 SBA-15的孔道结构与球形MCM-41的孔道结构相似,均为由球心向四周发散的结构,这使得只有当 TE M投射角度垂直于长程有序排列的孔道时,才有可能观测到孔道的有序性.但是,因为湿磨过程对 SBA-15的球形外貌有很强的破坏性,而破坏后的 SBA-15残片的形状、存在的位置都不可人为控制,这就直接导致了 TE M所观测的范围具有一定局限性.因而通过 TE M难以观测到破碎后的球形 SBA-15孔道的有序性.由图 12(d)可看到圆圈中分子筛的有序性,但范围较小,长程有序性较短,这与破碎的 SBA-15形状及观测的位置有一定的关系.
3 结 论
(1)本研究合成的球形MCM-41和球形 SBA-15具有典型的特征参数,比表面积、孔容积和孔径分别为1 107 m2·g-1和 583 m2·g-1,0.69 cm3· g-1和1.27 cm3·g-1,2.5 nm和 7.4 nm;
(2)湿磨过程对球形MCM-41的形貌基本没有影响,而球形 SBA-15经湿磨后其形貌很容易被破坏;
(3)合成的球形 SBA-15颗粒尺寸非常大,因此只有将球形 SBA-15湿磨破碎后,才可通过 TE M观测其内部有序性的孔道结构.这为观测大颗粒尺寸分子筛内部孔道结构提供了一种有效简便的操作手段.
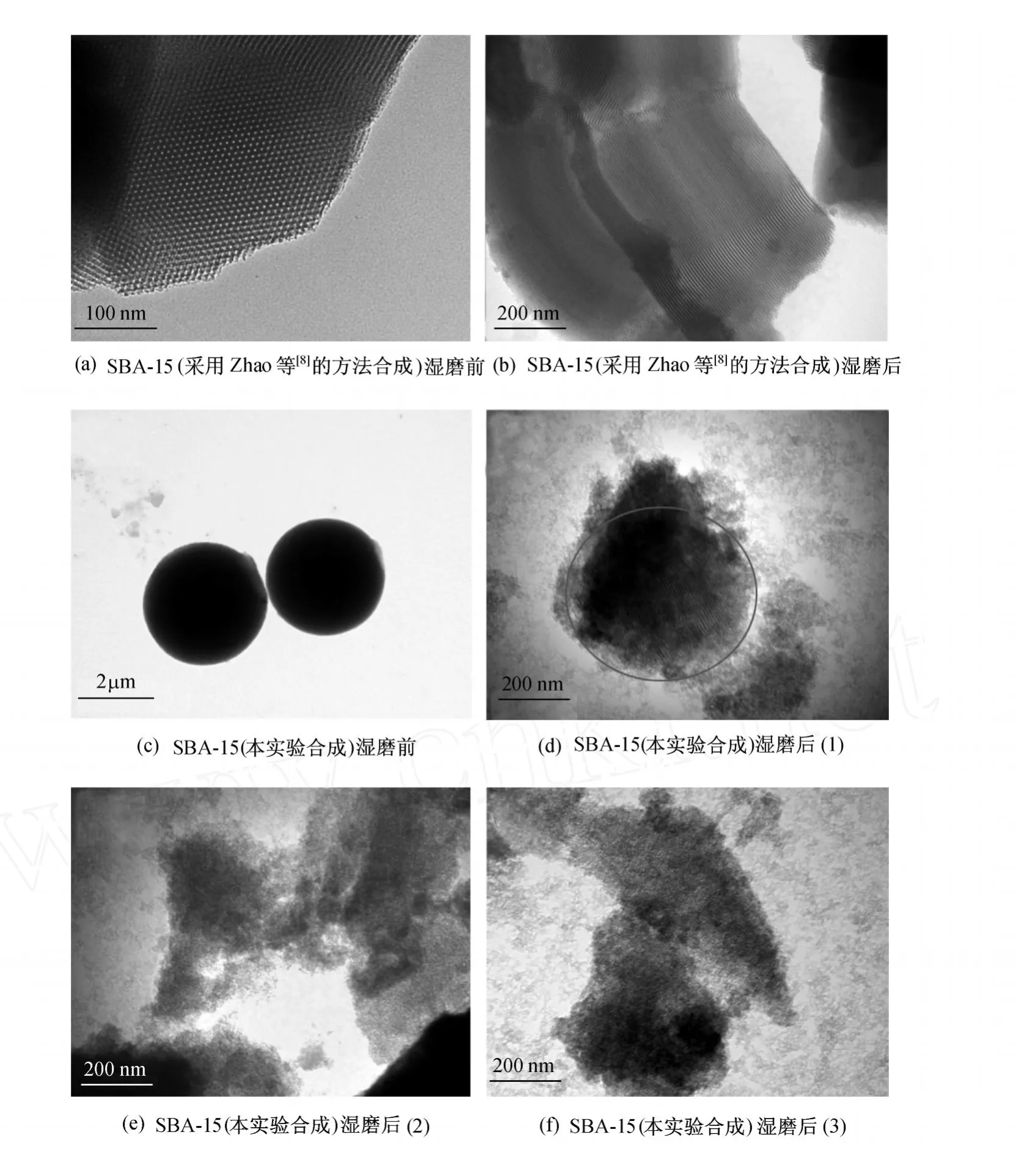
图12 湿磨过程对 SBA-15孔道结构的影响Fig.12 Effect of wet grinding on spherical SBA-15 pore structure
[1] KRESAEC T,LEONOW I CZM E,ROTHW J,et al. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanis m[J].Nature,1992, 359:710-712.
[2] BECKJ S,VARTUL IJ C,ROTHW J,et al.A new family of mesoporous molecular sieves prepared with liquid-crystal templates[J].J Am Chem Soc,1992, 114:10834-10843.
[3] KAPPPRS,BHATTACHARYYAA J.Ultrasound-triggered controlled drug delivery and biosensing using silica nanotubes[J].J Phys Chem C,2009,113(17):7155-7163.
[4] OJAN IR, RAOOF JB, FATHIS. Ferricyanide immobilized within organically modified MCM-41:application for electrocatalytic reduction of hydrogen peroxide[J].J Solid State Electrochem,2009,13(6):837-842.
[5] PORTELAR,CANELAM C,SANCHEZB. H2S photodegradation by TiO2/M-MCM-41(M=Cr or Ce):deactivation and by-product generation under UV-A and visible light[J].Appl Catal B,2008,84(3/4):643-650.
[6] PUANNGAMM,UNOB F. Preparationanduse of chemically modified MCM-41 and silica gel as selective adsorbents for Hg(Ⅱ)ions[J].J Hazard Mater, 2008,154:578-587.
[7] L IND S,ZHOUQ X.Effects of soil amendments on the extractability and speciation of cadmium, lead, and copper in a contaminated soil[J].Bull Environ Contam Toxicol,2009,83(1):136-140.
[8] ZHAOD,FENGJ,HUOQ,et al.Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores[J].Science,1998,279:548-552.
[9] XUEXM,LIF T.Removal of Cu(Ⅱ)from aqueous solution by adsorption onto functionalized SBA-16 mesoporous silica[J].MicroporousMesoporousMater, 2008,116:116-122.
[10] IZQU IERDO-BARBAI,SOUSAE,DOADR IOJ C,et al. Influence of mesoporous structure type on the controlled delivery of drugs:release of ibuprofen from MCM-48, SBA-15 and functionalized SBA-15[J].J Sol-Gel Sci Technol,2009,50(3):421-429.
[11] HOUY Z,JIX T,L IUG,et al. Immobilization of palladium in N-heterocyclic carbene functionalized SBA-15 for the catalytic application in aerobic oxidation of benzyl alcohol[J].Catal Commun,2009,10(10):1459-1462.
[12] ZHOUP,MENGQ T,HEG J.Highly sensitive fluorescence probe based on functional SBA-15 for selective detection of Hg2+in aqueous media[J].J EnvironMonit,2009,11(3):648-653.
[13] HANP,ZHANGHM,Q IUX P,et al.Palladium within ionic liquid functionalized mesoporous silica SBA-15 and its catalytic application in room-temperature Suzuki coupling reaction[J].J Mol Catal A:Chem,2008, 295(1/2):57-67.
[14] SHAHP,SR I DEV IN,PRABHUNEA,et al.Structural features of Penicillin acylase adsorption on APTES functionalized SBA-15[J].Microporous Mesoporous Mater,2008,116:157-165.
[15] YUC C,ZHANGL X,Q INF,et al.Composite in application of heavymetals ions adsorption[J].J Inorg Mater,2008,23(6):1231-1235.
[16] GRÜNM,UNGERK K,MATSUMOTOA,et al.Novel pathways for the preparation of mesoporous MCM-41 materials:control of porosity and morphology[J]. MicroporousMesoporousMater,1999,27:207-216.
[17] X IANB,L IU H,SUN Y C,et al. Preparation of spherical large-particleMCM-41 with abroad particle-size distribution by a modied pseudomorphic transformation [J].Microporous Mesoporous Mater,2009,121:73-78.
[18] KATIYARA,YADAV S,SM I RN IOTISP G,et al. Synthesis ofordered large pore SBA-15 spherical particlesforadsorption ofbiomolecules [J]. J ChromatogrA,2006,1122:13-20.
[19] MAY,QIL,MAJ M,et al.Large-pore mesoporous silica spheres:synthesis and application in HPLC[J]. Colloids SurfA,2003,229:1-8.
[20] 徐如人,庞文琴.分子筛与多孔材料化学[M].北京:科学出版社,2005:597-615.
[21] ZUKALA,THOMMESM,CˇEJKAJ,et al.Synthesis of highly ordered MCM-41 silica with spherical particles [J].Microporous Mesoporous Mater,2007,104:52-58.
[22] PAUWELSB,TENDELOOG V,THOELENC,et al. Structure determination of spherical MCM-41 particles [J].AdvMater,2001,13(17):1317-1320.
[23] LI USQ,COOLP,COLLARTO,et al.The influence of the alcohol concentration on the structural ordering of mesoporous silica:cosurfactant versus cosolvent[J].J Phys Chem B,2003,107:10405-10411.
