幽门螺杆菌 NCTC11637、NCTC11639毒素相关基因A的克隆、序列分析及真核表达载体的构建*
2010-01-24周建奖单可人
汪 苏,周建奖,单可人,赵 燕,谢 渊
1994年世界卫生组织国际癌症研究机构(IARC)正式将幽门螺杆菌(Helicobacter pylori,H p)列为Ⅰ类生物致癌因子。大量研究表明,幽门螺杆菌是慢性胃炎和消化性溃疡的致病菌,也是胃癌的致病因子之一,H p的致病机理仍不清楚。幽门螺杆菌分泌许多毒力蛋白,其中毒素相关基因A(cytotoxin-associated gene A,CagA)表达的蛋白CagA得到特别的关注。许多幽门螺杆菌菌株都能分泌CagA,CagA广泛存在于60%~70%的H p菌株中。许多流行病学研究也表明CagA阳性的H p菌株感染同萎缩性胃炎、十二指肠疾病和胃癌之间有显著相关。本实验以H p国际标准株NCTC11 637、NCTC11 639的基因组 DNA为模板进行PCR。PCR产物采用 TA法克隆CagA全长基因,测序并进行序列分析。根据测序结果选择PstI、B amHI两个酶切位点进行双酶切,连入真核表达载体pcDNA3.1/ZEO(-)中构建了CagA真核表达载体pcDNA3.1ZEO(-)/CagA7和pcDNA 3.1 ZEO(-)/CagA9。
1 材料与方法
1.1 幽门螺杆菌国际标准菌株 NCTC11 637、NCTC11 639由中国幽门螺杆菌菌株管理与保藏中心提供;pMD18-T原核克隆载体购于 TaKaRa公司,pcDNA3.1/ZEO(-)真核表达载体购于 Invitrogen公司;CagA的PCR引物由上海生工生物工程技术服务有限公司合成。
1.2 幽门螺杆菌CagA基因片段的制备 采用 PCR的方法制备CagA基因片段:(1)幽门螺杆菌基因组DNA的制备:幽门螺杆菌NCTC11637、NCTC11639于哥伦比亚血琼脂37℃微需氧培养3d,提取菌株DNA作为 PCR模板。(2)CagA基因片段的 PCR制备:上游引物:5′-AC AAT GAC TAA CGA AAC CA-3′;下 游 引 物 :5′-TTT TGG TAT TCC TTA ATC CT-3′〔1〕。以上述模板 ,采用高保真酶,重复30次循环,PCR产物以1%琼脂糖凝胶电泳,约3 500bp条带即为目的DNA。
1.3 CagA基因的原核载体的构建-TA克隆 CagA PCR产物1%琼脂糖凝胶电泳,切胶回收CagA片段,并在3′末端加A。将CagA加A产物和pMD18-T原核质粒16℃连接过夜,转化感受态大肠杆菌DH5α(CaCl2法制备),涂布于含氨苄青霉素的LB琼脂平板,37℃培养过夜。挑选转化子,置于LB培养液37℃培养过夜,通过煮沸裂解法提取质粒初步鉴定阳性重组克隆,并进行CagAPCR扩增,1%琼脂糖电泳,约有3 500bp条带者疑为阳性克隆,阳性克隆分别命名为 pMD18-T/CagA7和 pMD18-T/CagA9。送TaKaRa公司测序,并根据测序结果选择PstI、Bam HI两个位点进行双酶切鉴定。
1.4CagA基因真核载体构建 pMD18-T/CagA7和pMD18-T/CagA9分别进行PstI、BamHI双酶切1%琼脂糖凝胶电泳后,切取CagA目的基因进行切胶纯化。真核表达载体pcDNA3.1/ZEO(-)同样分别选取PstI、Bam HI两个位点酶切后进行切胶纯化。将目的基因和酶切纯化的pcDNA3.1/ZEO(-)16℃连接过夜,转化感受态大肠杆菌DH5α,涂布于含氨苄青霉素的LB琼脂平板,37℃培养过夜。用煮沸裂解法提取质粒初步判定为阳性克隆后,挑取阳性克隆,用双酶切和CagAPCR扩增鉴定阳性克隆。分别命名为pcDNA3.1ZEO(-)/CagA7、pcDNA3.1 ZEO(-)/CagA9。
1.5 pcDNA3.1ZEO(-)/CagA转染胃癌细胞 人胃癌细胞株AGS购于中科院上海细胞库,常规培养24h后用构建的pcDNA3.1ZEO(-)/CagA真核表达载体转染,根据 Invitrogen公司Lipofectamine 2000转染试剂说明书操作,48h后收集细胞,提取RNA和蛋白进行鉴定。
1.6 Western blot检测CagA蛋白的表达 提取细胞蛋白,SDS聚丙烯酰胺凝胶电泳分离后,电转移至 PVDF膜,膜在5%封闭液中室温孵育1h后加入一抗(兔多抗CagA,1∶500稀释)4℃过夜,TBST洗膜3次,每次10min,再加入 HRP标记的二抗,室温孵育膜2h,TBST洗膜2次,每次15min,TBS洗膜1次,15min;以 GAPDH作为对照,ECL室温孵育5min,胶片曝光显影。
1.7 细胞总RNA的提取及cDNA逆转 细胞采用 Trizol提取总 RNA,取 2000 ng的 mRNA,加入 Oligo dT18(10 pmol/μL)混匀后放置65℃水浴5min,迅速取出转置冰上,加入 dNTPs(10 mmol/μL)2.5μL;5 ×buffer 5μL;MMV(200 U/μL)1μL;RNA 酶抑制剂 (40 U/μL)0.5μL;加DEPC处理水至25μL。进行逆转录反应,反应条件:37℃60 min,75℃60 min,所得cDNA于-20℃保存。根据文献设计 CagA 引物〔4〕。上游引物:5′-AAT ACA CCA ACG CCT CCA AG-3′;下游引物 :5′-TTG TTG CCG CTT TTG CTC TC-3′。PCR产物以1.5%琼脂糖凝胶电泳,约397bp条带即为目的DNA。
1.8 实时荧光定量 PCR 胃泌素及β-actin检测试剂由ABI公司提供,按说明书操作。反应条件:50℃2min,95℃10min,95℃15s,60℃1min,40个循环,数据采用 ABI7300 SDS Software分析。
1.9 相对mRNA表达水平的计算 采集胃泌素及β-actin基因各循环荧光信号,用 SDS1.4软件计算Ct值、△△Ct值及相对表达值 RQ〔5〕,△△Ct=(Ctgastrin-Ctβ-actin)处理组 -(Ctgastrin-Ctβ-actin)对照组 ,RQ=2-△△Ct。
1.10 统计处理 数据以均数±标准差表示,采用 SPSS 11.5软件进行单因素方差分析,P<0.05为差异有显著性。
2 结 果
2.1 NCTC11 637、NCTC11 639CagA基因片段的制备 提取NCTC11 637菌株及NCTC11 639菌株DNA后,用CagA引物进行 PCR扩增,1%琼脂糖凝胶电泳,在约3 500bp处得到CagA目的条带,见图1A。
2.2 pMD18-T/CagA原核克隆载体的鉴定 用煮沸裂解法提取质粒,初步鉴定为阳性重组克隆后,用质粒小提试剂盒提取阳性克隆。用CagA引物进行PCR扩增,均得到约3 500bp的目的条带,见图1B,测序后选用PstI、B amHI进行双酶切鉴定,见图1C,在2 500bp~5 000bp之间得到两条条带。上方约3 500bp处为CagA目的片段,而下方2692bp处为pMD18-T原核载体。表明pMD18-T/CagA7和pMD18-T/CagA9原核克隆载体构建成功。
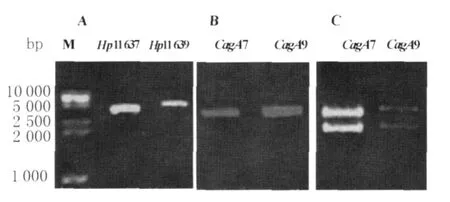
图1 原核载体pMD18-T/CagA的构建和鉴定A:幽门螺杆菌 NCTC11 637和 NCTC11 639CagA PCR扩增电泳图;B:原核载体pMD18-T/CagA CagA PCR鉴定电泳图;C:原核载体pMD18-T/CagA PstI、Bam HI双酶切鉴定电泳图;M:marker;Hp11 637:幽门螺杆菌 NCTC11 637;Hp11 639:幽门螺杆菌NCTC11 639;CagA7:原核载体pMD18-T/CagA7;CagA9:原核载体pMD18-T/CagA9Fig.1 Construction and identification of prokaryotic vector pMD18-T/CagA
2.3 pcDNA3.1ZEO(-)/CagA7和pcDNA 3.1 ZEO(-)/CagA9真核表达载体鉴定 用煮沸裂解法提取质粒初步鉴定为阳性重组克隆后,用CagA引物进行 PCR扩增,均得到约3 500bp的目的条带,见图2A。同时用PstI、B amHI进行双酶切鉴定,见图2B,在2 500bp~5 000bp之间得到两条条带,其中上方5 000bp处为pcDNA3.1/ZEO(-)载体,下方约3 500bp处为CagA基因。结果表明幽门螺杆菌pcDNA3.1ZEO(-)/CagA7和pcDNA3.1ZEO(-)/CagA9真核表达载体构建成功。
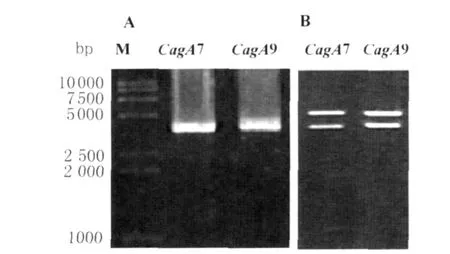
图2 真核表达载体pcDNA3.1/ZEO(-)/CagA的鉴定A:真核表达载体pcDNA3.1/ZEO(-)/CagA CagA PCR鉴定电泳图;B:真核表达载体pcDNA3.1/ZEO(-)/CagA PstI、Bam HI双酶切鉴定电泳图;M:marker;CagA7:真核表达载体 pcDNA3.1/ZEO(-)/CagA7;CagA9:真核表达载体 pcDNA3.1/ZEO(-)/CagA9Fig.2 Identification of eukaryotic vector pcDNA3.1/ZEO(-)/CagA
2.4CagA基因序列测定和同源性分析 测序结果显示:NCTC11 637中CagA基因全长为 3 467bp,NCTC11 639CagA基因全长为 3 594bp。将测序结果登录 GenBank,登录号分别为 GQ161 098(NCTC11 637)和 GQ161 099(NCTC11 639)。NCTC11 637CagA的序列与 GenBank收录的NCTC11 637CagA序列(AB015 416)基本相符,核苷酸序列同源性达99.22%。NCTC11 639CagA全序列 GenBank未见收录,而 NCTC1 1639CagA与本实验构建的NCTC11 637序列比对同源性为87.88%。
2.5 CagA蛋白酪氨酸磷酸化位点 CagA蛋白酪氨酸磷酸化位点在CagA基因的 EPIYA(Glu-Pro-Ile-Tyr-Ala)模体上。根据pMD18-T/CagA7和pMD18-T/CagA9的测序结果,利用软件分析 CagA蛋白的 EPIYA位点,显示 NCTC11 637和NCTC 11 639CagA蛋白酪氨酸磷酸化位点各有两个,分别为 NCTC11 637 EPIYA-A、-B和 NCTC11 637 EPIYA-A、-B。
2.6 pcDNA3.1ZEO(-)/CagA转染胃癌细胞AGS后CagA的表达 将构建的真核表达载体pcDNA3.1ZEO(-)/CagA转染胃癌细胞株AGS 48h后提取细胞总RNA和细胞蛋白,分别用RTPCR和Western-blot检测CagA mRNA和蛋白的表达,结果显示转染后AGS细胞表达CagA,见图3,表明pcDNA3.1/ZEO(-)/CagA真核表达质粒转染成功。
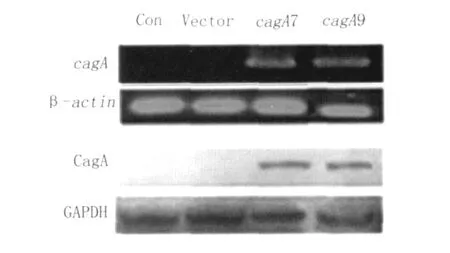
图3 pcDNA3.1/ZEO(-)/CagA转染胃癌细胞AGS后CagAmRNA(上部)和蛋白(下部)表达Con:未转染的细胞对照;Vector:空载体转染的细胞对照;CagA7:pcDNA3.1/ZEO(-)/CagA7转染;CagA9:pcDNA3.1/ZEO(-)/CagA9转染Fig.3 Expression of CagA mRNA(upper)and protein(down)in gastric cancer AGS cells transfected with pcDNA3.1/ZEO(-)/CagA
2.7 pcDNA3.1ZEO(-)/CagA转染细胞后胃泌素mRNA表达 pcDNA3.1ZEO(-)/CagA转染AGS细胞后,以对照组胃泌素 mRNA的表达量为对照,各转染组中胃泌素mRNA的相对表达量分别为:4.86±0.45(pcDNA3.1)、10.56±0.87(pcDNA3.1/CagA7)及10.63±0.84(pcDNA3.1/CagA9),见图4。结果显示CagA的转染显著增加了AGS细胞中胃泌素基因的表达(P<0.05)。
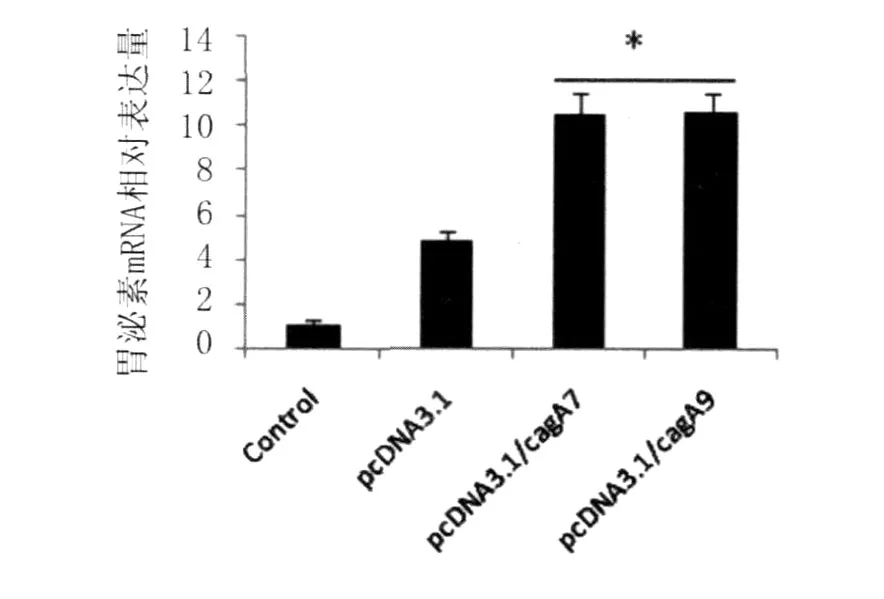
图4 pcDNA3.1ZEO(-)/CagA转染 AGS细胞后胃泌素mRNA表达Control:未转染的细胞对照;pcDNA3.1:空载体转染的细胞对照;pcDNA3.1/CagA7:pcDNA3.1/ZEO(-)/CagA7转染;pcDNA3.1/CagA9:pcDNA3.1/ZEO(-)/CagA9转染;*:与空载体转染组比差异有统计学意义 (P<0.05)Fig.4 The levels of gastrin mRNA in AGS cells transfected with the pcDNA3.1ZEO(-)/CagA
3 讨 论
CagA基因编码120~145×103Da的CagA蛋白,通过Ⅳ分泌系统输送到胃上皮细胞,然后定位于细胞的浆膜面,转位的CagA通过Src家族蛋白激酶(SFK)发生酪氨酸磷酸化后能与多种细胞蛋白作用,调控细胞生长和运动相关的信号通路,使细胞骨架重排、运动力增强,转录因子表达改变,导致细胞增殖〔2〕。磷酸化的CagA是其主要的活性形式,而CagA中酪氨酸磷酸化的位点主要发生在 EPYIA模体上。所以CagA的磷酸化水平与 EPIYA模体的数量相关。NCTC11 637和NCTC11 639为西方菌株,一些学者的研究发现NCTC11 637的CagA有 EPIYA-A、-B、-C、-C、-C 五个酪氨酸磷酸化位点〔3〕,但是我们的研究发现 NCTC11 637的 CagA仅在羧基端有两个酪氨酸磷酸化位点,即 EPIYAA、-B,也有日本学者发现,NCTC11 637其羧基端只有两个酪氨酸磷酸化位点〔4〕,与我们序列的同源性高达99.25%(AB015 416),因此我们认为CagA基因的异质性可能与它的流行地区有关。而NCTC11 639的序列在 GenBank中未见收录,我们将NCTC11639和NCTC11637测序序列比对发现两者的同源性有87.88%,并且NCTC11 639也只有两个酪氨酸磷酸化位点 EPIYA-A和 EPIYA-B。虽然有研究表明,分子量越大、磷酸化位点越多的CagA与H p菌株毒力高低和胃癌的关系更密切〔5〕,但是目前的流行病学研究发现,胃癌的发病率在中国、日本等亚洲国家逐年增加,而欧美国家逐年下降,这种差异是否与不同地区的流行菌株和CagA的酪氨酸磷酸化位点有关尚待进一步研究。
本研究采用T-A克隆法将PCR产物直接克隆到原核载体pMD18-T上,使用高保真酶确保了产物扩增的正确性。因为高保真酶的特殊性,产物需要在3′端加入一非模板来源的突出A。突出A则可与 T克隆载体的5′突出的 T互补配对,并被连接酶连接形成重组体。原核克隆载体构建成功后,根据测序后限制性核酸内切酶谱分析结果选择单一酶切位点的两个限制性核酸内切酶,双酶切原核克隆载体后连入真核表达载体pcDNA3.1/ZEO(-)中,成功构建了NCTC 11 637和NCTC 11 639 CagA全长真核表达载体pcDNA3.1ZEO(-)/CagA7和pcDNA3.1ZEO(-)/CagA9。随后我们将构建的真核表达载体转染胃癌细胞株A GS,并在AGS细胞中检测到了CagAmRNA和蛋白质的表达,并且发现AGS细胞中胃泌素基因的表达上调,说明幽门螺杆菌毒素相关蛋白 CagA调控了胃泌素基因的表达。
幽门螺杆菌毒素相关蛋白CagA和胃泌素是胃癌发生发展的两大重要因素,尽管过去的研究发现他们之间有一定的联系,但迄今为止,尚无直接研究他们相互关系的文献报道。而我们通过构建含CagA基因的真核表达载体转染胃癌细胞,用实时荧光定量PCR定量检测胃泌素基因表达水平的变化来直接研究它们之间的相互作用,因此我们的研究首次将两者直接联系起来,证实了CagA是调控胃泌素基因表达的相关蛋白。
〔1〕Zhou JC,Zhang JZ,Xu Cp,et al.Helicobacter pylorifull-length of CagA gene cloning and sequence analysis〔J〕.Journal of Third Military Medical Uneversity,2005,27(1):36-38.
〔2〕Masanori H.Helicobacter pyloriCagA-a bacterial intruder conspiring gastric carcinogenesis〔J〕.J Cancer,2006,119:217-1223.
〔3〕Johannes G,Arnoud H.M,Ernst K,et al.Pathogenesis ofHelicobacter pyloriinfection〔J〕.Clin Microbiol Revi,2006,19(3)9-490.
〔4〕Hoshino F,Katayama K,Watanabe K,et al.Heterogeneity found in the CagA gene ofHelicobacter pylorifrom Japanese and non-Japanese isolates〔J〕.J Gastroenterol,2000,35(12):890-897.
〔5〕Higashi H,Tsutsumi R,Fujita A,et al.Biological activity of theHelicobacter pylorivirulence factor CagA is determined by variation in the tyrosine phosphorylation sites〔J〕.Proc Natl Acad Sci U A,2002,99(22):14428-14433.
