密度泛函理论研究Mgn H2(n=2-12)团簇的结构和电子性质
2010-01-05闫红霞葛桂贤曹海宾
闫红霞,葛桂贤,井 群,曹海宾
密度泛函理论研究Mgn H2(n=2-12)团簇的结构和电子性质
闫红霞,葛桂贤,井 群,曹海宾
(石河子大学师范学院物理系/石河子大学师范学院生态物理重点实验室,石河子832003)
为了研究Mgn团簇和 H2的相互作用机制,采用密度泛函理论中的广义梯度近似(GGA)对Mgn团簇吸附H2的几何构型进行优化,通过能量计算寻找出Mgn团簇和MgnH2团簇的最低能量结构,并计算了对象最低能量结构的平均结合能、二阶能量差分、吸附能、垂直电离势、垂直亲和势和能隙。分析结果表明 H2分子的吸附对Mgn团簇的稳定性、成键特性和结构的影响均较小,说明Mgn团簇储氢具有易于吸放的可能性;二阶能量差分和吸附能表明Mg4H2和Mg10H2团簇是比较稳定的。
MgnH2团簇;几何结构;电子性质
为了保护环境,人们一直在致力于开发新能源。氢就是一种很理想的清洁能源,如何提高储氢材料的储氢量和降低材料成本,节约贵重金属是一个亟待解决的问题。金属储氢因其安全、高能、经济、技术和资源等优势受到人们的青睐。许多金属、合金以及类金属都被作为储氢材料来研究。另外,鉴于团簇具有不同于其组成分子和块体的奇异性质,很强的尺寸依赖性以及特殊的物理化学性质,人们对团簇的上述性质进行了研究,如 G Dietrich等[1]研究了钒团簇对氢的吸附,Hege Stromsnes等[2]研究了金团簇对氢的吸附,H Yukawa等[3]研究了掺杂了3d过渡金属的LaNi5团簇储氢的能力。
镁因其储氢容量大(7.6%)、质量轻和价格低廉而倍受关注[4-9],人们从理论和实验两个方面研究了镁团簇及其吸附氢的性能。Vijay Kumar等[10]采用分子动力学密度泛函理论中的局域密度近似模拟退火研究了Mgn(n=2~13)团簇的结构、生长以及结合的性质;而 P Delaly等[11]研究了Mgn(n≤20)团簇的成键性质随团簇尺寸的演变;Muneyuki Tsuda等[12]基于第一性原理的密度泛函理论研究了3d过渡金属对MgnH2团簇释放 H2的影响趋势,结果表明 Sc和 Ni的催化能力较强;D Chen等[13]采用第一性原理的密度泛函理论研究了过渡元素M对Mg H2的电子结构的影响,结果表明在纯的Mg H2团簇中,Mg和 H之间的主要作用为化学作用。然而,镁团簇和氢之间相互作用的机制尚不十分明确。
本文系统研究了氢分子的吸附对不同尺寸镁团簇的结构和电子性质的影响,为以后深入探讨镁吸附氢的原理提供参考。
1 计算方法
采用密度泛函理论(DFT)下的广义梯度近似(GGA),用DMoL 3软件包[14]对所能预见的全部构型进行结构优化和电子性质计算。在 GGA中,选择Perdew-Burke-Ernzerhof(RPBE)交换关联泛函。所有的计算均是在Fine网格下完成的,采用带极化的双数值原子基组(DNP))进行全电子计算,自洽过程以体系的能量和电荷密度分布是否收敛为依据,精度均优于10-5a.u.。梯度和位移的收敛精度优于10-2a.u./nm和10-4nm,能量的收敛精度优于2×10-5a.u.。另外,在缺省轨道占据拖尾效应参数下,对MgnH2(n=2-12)团簇的所有几何构型进行优化、能量和性质计算,在拖尾效应参数为0.005a.u.时计算其频率。为了对所用计算方法进行标定,用不同的泛函对Mg2,H2,Mg-H的结合能和键长进行了计算,不同计算方法的结果与实验值的对比见表1。
从表1可以看出,选择RPBE交换关联泛函计算的结果与实验值符合的很好,表明该方法很好地描述了Mg2和 H2二聚体,因此这种方法也适用于MgnH2(n=2~12)团簇。
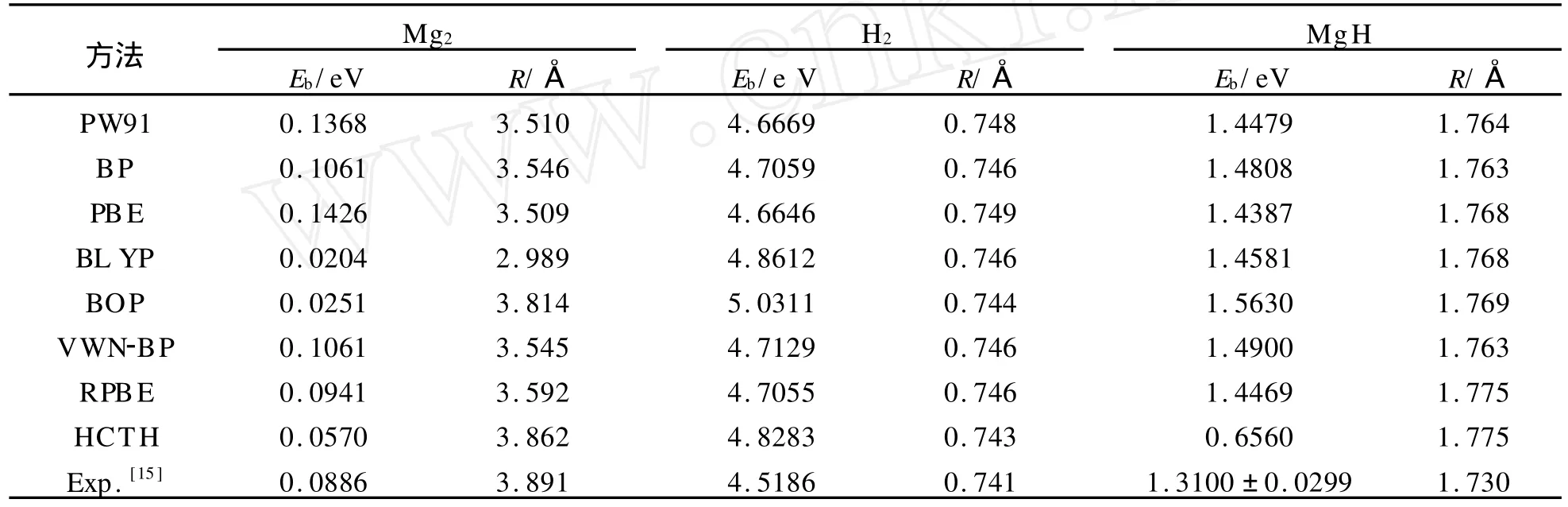
表1 不同计算方法Mg2,H2,MgH二聚体结合能和键长与实验值的对比Tab.1 Binding energy and bond length of Mg2,H2,MgH dimer calculate by different methods vs experimental results
2 结果与分析
2.1 几何结构
在几何优化的基础上对计算频率,把能量最低且振动频率为正值的结构确定为最低能量结构。为了寻找氢分子最可行的吸附位置,考虑Mg原子的顶部、空位和桥位3种不同的吸附位置,为了研究H2分子的吸附对镁团簇的影响,使用同样的泛函和基组对Mgn(n=2~12)团簇进行几何优化,得到镁团簇的幻数为 n=4、10,计算结果与文献[16-17]的结果一致。
图1给出了Mgn和MgnH2(n=2-12)团簇的最低能量结构。如图1中2b所示,H2吸附在Mg原子顶部的平面结构是Mg2H2团簇的最低能量结构,H2和Mg不在一条直线上。Mg3团簇的最低能量结构是三角形,H2分子吸附在桥位Mg3H2团簇能量最低,见图1中3b。Mg4团簇的最低能量结构是三角锥。空位上吸附 H2分子形成的Mg4H2团簇的结构能量最低,与Mg5团簇的最低能量结构相似,同为三角双锥。三角双锥的Mg5团簇的空位上吸附 H2形成的舟形Mg5H2团簇的能量最低,这个结构近似于Mg6团簇的最低能量结构。H2分子吸附在Mg6团簇桥位上形成最低能量结构的Mg6H2团簇,该结构与Mg7团簇的最低能量结构五角双锥相差很远,对 Mg-Mg键长影响也较大,见图 2。H2分子吸附在Mg7团簇的五边形的一个顶点上形成了Mg7H2团簇的最低能量结构。当n=8-12,MgnH2团簇的最低能量结构是 H2分子吸附在Mgn团簇的空位上生成的。
从上面的分析可以看出,H2分子的吸附基本没有改变Mgn团簇的最低能量结构。
为了进一步考察H2分子的吸附对Mgn团簇结构的影响,计算Mgn团簇和Mg2H2团簇中 Mg-Mg键的平均键长随团簇尺寸的变化曲线,结果见图2。
从图2可知,除了 n=6,7外,H2分子的吸附基本没有改变Mg-Mg键的平均键长,表明在大多数情况下H2分子的吸附对Mgn团簇的稳定性影响较小。在n=6,7时,Mgn团簇的最低能量结构由三角双锥到五角双锥过渡,H2分子的影响略有起伏(图 1)。
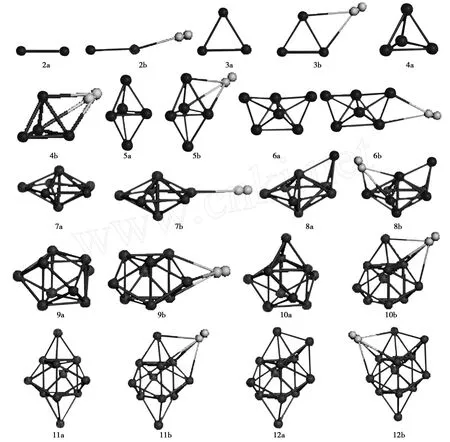
图1 Mgn和Mgn H2(n=2-12)团簇的最低能量结构Fig.1 Lowest energy structures of Mgn and Mgn H2(n=2-12)clusters
2.2 稳定性和电子性质
平均结合能能很好地描述团簇的稳定性。Mgn和MgnH2(n=2-12)团簇的平均结合能的计算公式如下:
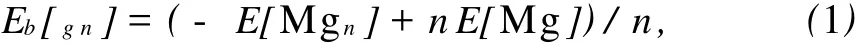
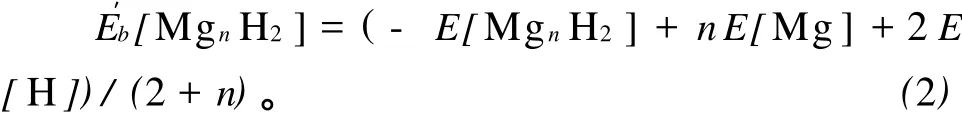
式(1)、(2)中 E[Mgn],E[Mg],E[MgnH2],E[H]分别为最稳定结构的Mgn,Mg,MgnH2,H的总能量。
图2b是Mgn和MgnH2(n=2-12)团簇的平 均结合能随团簇尺寸变化的规律。
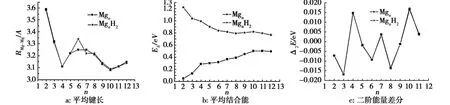
图2 Mgn和Mgn H2团簇最低能量结构的稳定性Fig.2 The stability of ground state Mgn and Mgn H2(n=2-12)clusters
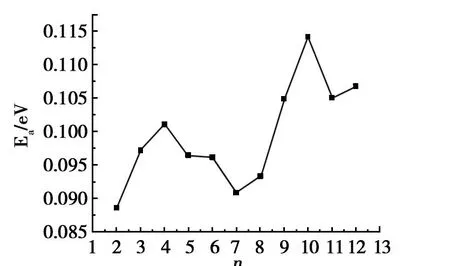
图3 Mgn H2团簇最低能量结构的吸附能Fig.3 The adsorption energies of Mgn H2(n=2-12)clusters
从图2b可以看出,MgnH2团簇的平均结合能大于Mgn团簇的平均结合能,这可能由MgnH2团簇中H2中的2个 H原子之间的氢键所致。MgnH2团簇的平均结合能随着团簇尺寸的增加而减小,最终趋于一个固定的值。说明随着团簇尺寸的增大,氢键对MgnH2团簇的平均结合能的影响逐渐减弱。
二阶能量差分是很好的理解团簇相对稳定性的一个物理量,其计算公式如下:
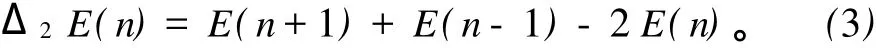
式(3)中 E(n)是团簇总的结合能。
图2c给出了Mgn和MgnH2(n=2-12)团簇的二阶能量差分。从图2c可以看出,镁团簇的幻数为4和10,这与文献[11]符合得很好。Mgn和MgnH2二阶能量差分的值相差极小,且随团簇尺寸的变化趋势相同,这表明吸附 H2分子对镁团簇的成键特性影响较小。
吸附能是反映H2和Mgn团簇相互作用能力的物理量,吸附能 Eadv的计算公式为:
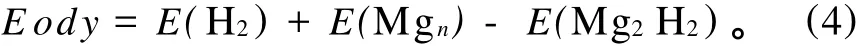
图3反映了Mg4H2团簇最低能量结构的吸附能随团簇尺寸的变化。
由图3可见,在Mg4H2和Mg10H2处出现了峰值,这表明Mg4H2和 Mg10H2是稳定的团簇,这与二阶能量差分的结果一致。另外,吸附能很小(0.09~0.11 eV),说明 H2和Mgn介于物理吸附和化学吸附之间[18],作用力较弱,也证明了MgnH2团簇的平均结合能大于Mgn团簇的平均结合能,基本上由H2中的氢键所致。这样MgnH2团簇既有较好的稳定性,又较容易释放出 H2。
电离势和亲和势也是反映团簇稳定性的物理量。团簇的电离势是指一个中性团簇失去一个电子的结合能,可以通过计算中性团簇基态能量和阳离子基态能量之差得到。垂直电离势的值越大,表明该团簇越稳定,团簇的亲和势是指一个中性团簇得到一个电子的结合能,可以通过计算中性团簇基态能量和阴离子基态能量差得到。垂直亲和势的值越小,团簇越稳定。从图7中可以看出,垂直电离势VIP和垂直亲和势VEA的定义分别如下:

图4a给出了Mgn和MgnH2团簇最低能量结构的垂直电离势。从图6可以看出,吸附 H2分子对镁团簇的稳定性影响较小。
图4b给出了团簇的垂直亲和势随团簇尺寸的变化规律,吸附H2分子对镁团簇的稳定性影响较小。
图4c给出了团簇的最高已占据轨道与最低未占据轨道的能隙 Eg随团簇尺寸的变化规律。能隙Eg反映了电子从占据轨道向未占据轨道发生跃迁的能力,在一定程度上代表分子参与化学反应的能力。从图8可以看出,吸附 H2分子对镁团簇的化学活性影响很小。
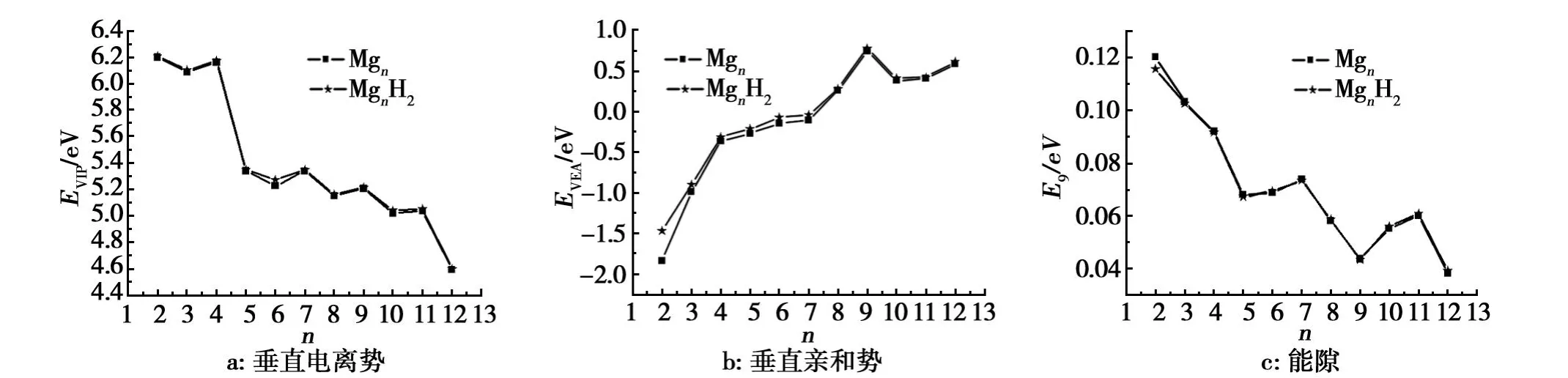
图4 Mgn和Mgn H2团簇最低能量结构的电子性质Fig.4 The Electronic Properties of ground state Mgn and Mgn H2(n=2-12)clusters
3 结论
1)通过与镁团簇相比,H2分子的吸附对镁团簇的成键特性和结构的影响比较小,H2分子的吸附基本没有改变镁团簇的稳定性。
2)通过吸附能发现Mgn团簇易于吸放氢气。3)二阶能量差分和吸附能表明Mg4H2和Mg10H2团簇是稳定的。
致谢:感谢河南大学物理与电子学院提供的计算软件。
[1]GDietrich,K Dasgupta,K Lützenkirchen,et al.Chemisorption of hydrogen on a V5+cluster[J].Chemical Physics Letters,1996,252(1-2):141-146.
[2]Hege Stromsnes,Sutjano Jusuf,Bernd Schimmelpfennig,et al.A theoretical study of the chemisorption of molecular hydrogen on a seven atom gold cluster[J].Journal of Molecular Structure,2001,567-568(2-3):137-143.
[3]H Yukawa,M Moringa,Y Takahashi.Alloying effect on the electronic structures of hydrogen storage compounds[J].Journal of Alloys and Compounds,1997,253-254(1-2):322-325.
[4]Pieter C M M Magusin,W Peter Kalisvaart,Peter H L Notten,et al.Hydrogen sites and dynamics in lightweight hydrogen-storage material magnesium--scandium hydride investigated with 1H and 2H NMR[J].Chemical Physics Letters,2008,456(1-3):55-58.
[5]A L Companion.Interaction of hydrogen atoms with small magnesium clusters doped with aluminum and 3d transition metals[J].Journal of the Less-Common metals,1984,98(1):121-130.
[6]A S Pedersen,J Kjoller,B Larsen,et al.Magnesium for hydrogen storage[J].Int.J.Hydrogen Energy,1983,8(3):205-211.
[7]Sharon Broude,R Benny Gerber.Solvation of metal atoms in quantum clusters:structural and vibrational properties of Hg(H2)12 and Mg(H2)12[J].Chemical Physics Letters,1996,258(3-4):416-420.
[8]葛桂贤,孙茂珠,殷保祥.密度泛函理论对NiBen(n=2-12)团簇结构和性质的研究[J].石河子大学学报:自然科学版,2008,26(6):781-785.
[9]Jian-Min Zhang,Ying-Ni Duan,Ke-Wei Xu,et al.Ab initio calculation of neutral and singly charged Mgn(n≤11)clusters[J].Physica B,2008,403(18):3119-3124.
[10]Vijay Kumar,Roberto Car.Structure,growth,and bonding nature of Mg clusters[J].Physical Review B,1991,44(15):8243-8255.
[11]PDelaly,PBallone,J Buttet.Metallic bonding in magnesium microclusters[J].Physical Review B,1992,45(7):3838-3841.
[12]Muneyuki Tsuda,Wilson Agerico Dino,Hideaki Kasai,et al.Mg-H dissociation of magnesium hydride Mg H2catalyzed by 3d transition metals[J].Thin Solid Films,2006,509(1-2):157-159.
[13]D Chen,YM Wang,L Chen,et al.Alloying effectsof transition metals on chemical bonding in magnesium hydride MgH2[J].Acta Materialia,2004,52(2):521-528.
[14]Delley B.From molecules to solids with the DMol3 approach[J].J Chem Phys,2000,113(18):7756-7764.
[15]Fernando Ruette,Morella Sánchez,Rafael Aňez,et al.Diatomic molecule data for parametric methods.I[J].Journal of Molecular Structure:Theochem,2005,729(1-2):19-37.
[16]Julius Jellinek,Paulo H Acioli.Magnesium clusters:structural and electronic properties and the size-induced nonmetal-to-metal transition[J].J Phys Chem A,2002,106(45):10919-10925.
[17]Andrey Lyalin,Ilia A,Solov′yov,et al.Evolution of the electronic and ionic structure of Mg clusters with increase in cluster size[J].Physical Review A,2003,67(6):063203-1-063203-13.
[18]赵振国.吸附作用应用原理[M].北京:化学工业出版社,1992,64-64.
Density Functional Theory Study of the Structure and Electronic Properties of MgnH2(n=2-12)Clusters
YAN Hongxia,GE Guixian,JING Qun,CAO Haibin
(Department of Physics,Normal College/Key laboratory of Ecophysics,Shihezi University,Shihezi 832003,China)
In order to study the principle of magnesium hydride,geometric structures of Mgnand MgnH2(n=2-12)clusters are optimized by using the generalized gradient approximation density f unctional theory.Energy and electronic properties have been calculated.In contrast with Mgnclusters,it is found that the adsorbed hydrogen molecule affects little for the structure,the stability of magnesium clusters and electron properties of Mgnclusters.Mg4H2and Mg10H2clusters are shown to be more stable than neighboring ones by the investigations on the second finite difference and absorb energy.
MgnH2cluster;geometries;electronic properties
O641
A
1007-7383(2010)04-0524-05
2009-11-06
闫红霞(1982-),女,讲师,从事储氢材料研究;e-mail:yanhongxia@shzu.edu.cn。
