伪狂犬病毒TK基因缺失通用转移载体的构建及初步应用
2009-11-11韩静芳陈瑞爱邵定勇唐满华梁桂益
韩静芳 陈瑞爱 邵定勇 唐满华 梁桂益
摘要以伪狂犬病毒为载体表达其他病原体抗原蛋白是伪狂犬病疫苗研究的一个重要方向。根据已发表的伪狂犬病毒(PRV)Ra株TK基因序列设计2对引物,用PCR方法得到同源左右臂,克隆入PUC19载体中。利用平末端连接的方法将绿色荧光蛋白(EGFP)的基因表达盒插入到缺失位点,并在下游引入1个多克隆位点,构建缺失通用转移载体PUC19TK-EGFP。用脂质体转染试剂盒将PUC19TK-EGFP和PRV-Ra株的基因组共转染BHK细胞,以EGFP为标记基因,用噬斑法得到纯化的重组病毒,命名为TK/EGFP,为研制以伪狂犬病毒为载体的二价或多价基因工程疫苗奠定了物质基础。
关键词伪狂犬病毒; TK基因;通用转移载体
中图分类号 S858.292 文献标识码A文章编号 1007-5739(2009)15-0308-03
ConstructionandPrimaryApplicationofUniversalTransferVectorDeletingTK ofPseudorabiesVirus
HAN Jing-fangCHEN Rui-ai*SHAO Ding-yongTANG Man-huaLIANG Gui-yi
(Guangdong Dahuanong Animal Health Products Co., Ltd., XinxingGuangdong 527400)
AbstractUse of PRV as a system for expression of foreign antigens has been increasingly evaluated as an alternative to conventional vaccine production system. In this study,according to the Genbank open sequence,two pairs of primer were designed in order to obtain homology recombination arms of TK gene of PRV-Ra strain. Then both of them were inserted into PUC19 vector. A report gene expression cassette,EGFP containing a multicloning sites,was inserted into PUC19HEK/TK to construct a universal transfer vector PUC19TK-EGFP. It was used to co-transfect BHK cell together with the genome of PRV Ra strain,a recombinat virus was selected and purified 3 times in BHK cell through EGFP gene and named TK/EGFP. The above results demonstrated that the universal transfer vector might be contributive to developed bivalence or multivalence genetically engineering vaccine in the future.
Key wordsPseudorabies virus ;TK gene;universal transfer vector
伪狂犬病(Pesudorabies, PR)是由伪狂犬病病毒(Pesudo-rabies virus,PRV)引起的多种家畜和野生动物的一种急性传染病,是严重危害全球养猪业的传染病之一[1]。 PRV属于疱疹病毒科(Herpesviridac)、α-疱疹病毒亚科(Alphaher-pesririnae)水痘病毒属(Varicello virus)的疱疹病毒Ⅰ型(Porcine herpesvirus TypeⅠ),其基因组为双链DNA,全长约150kb,编码72~100种蛋白质,存在许多与病毒复制无关的非必需基因,在这些非必需基因中能够插入其他病原的免疫原性基因或报告基因而不影响病毒的增殖。PRV的这种特性为其作为病毒活载体用于构建二价或多价基因工程疫苗提供了分子操作基础[2]。病毒的胸苷激酶基因是PRV的主要毒力基因,也是病毒的非必需基因,将该基因缺失不影响病毒在细胞中的增殖,但其毒力大大降低[3]。本研究用PCR方法扩增了TK基因的左右臂TKL和TKR,并克隆到PCU19载体中,这样在TKL和TKR基因之间缺失了203bp,用平末端连接法在此位点插入带CMV启动子和多克隆位点的EGFP报告基因,构建通用转移载体,并用蚀斑法得到纯化的缺失重组病毒TK-/EGFP。由于在EGFP的下游引入1个多克隆位点,这为将来构建以伪狂犬病毒为载体基因工程苗奠定基础。
1材料与方法
1.1材料
1.1.1病毒与细胞系。伪狂犬病毒(PRV)Ra株购自中国兽药监察所,BHK-15细胞由广东大华农生药研发中心保存。
1.1.2质粒与菌株。载体pcDNA 3.0(含EGFP基因)、载体PUC19以及大肠杆菌DH5α均由广东大华农生药研发中心保存。
1.1.3工具酶与试剂。LA Taq DNA聚合酶、XbaⅠ、KpnⅠ等多种限制性内切酶、T4连接酶、Klenow末端补平酶以及DNA Marker DL 2000等均购自大连宝生物工程有限公司;质粒回收试剂盒、DNA胶回收试剂盒购自上海生物工程公司;脂质体Lipofectamine 2000为Invitrogen公司产品。

1.1.4培养基与血清。细胞基础培养基DMEM和小牛血清为GIBCO公司产品;细菌培养用LB培养基和琼脂粉均购自广州美津生物技术有限公司。
1.1.5PCR引物设计。参照Genbank上发表的PRV-Ra株的TK基因序列合成2对引物用以扩增左右重组臂(TKL 、TKR分别为959bp和977bp),由上海生物工程公司合成。
P-TKLs:5′-ATTGGTACCGCTGCTCGTCCACCTC GG-3′(KpnⅠ)
P-TKLa:5′-GTCTCTAGAAGTACGCCATCGGCTC GG-3′(XbaⅠ)
P-TKRs:5′-GTCTCTAGAGTCTTTGACCGCCACC CG-3′(XbaⅠ)
P-TKRa:5′-ATTGCATGCCCGACCAGGACGAACA GG-3′(SphⅠ)
合成引物Ps、Pa,用以检测重组病毒中TK缺失基因,大小约为600bp,由上海生物工程公司合成。
Ps:5′-GCGTTCGTAGAAGCGGTTGTG-3′
Pa:5′-GTGTTGACCAGCATGGCGTAGA-3′
合成引物Ps-EGFP、Pa-EGFP,用以检测重组病毒中EGFP基因,大小为206bp,由上海生物工程公司合成。
Ps-EGFP:5′-GGGAGGATTGGGAAGACA-3′
Pa-EGFP:5′-AAGGGAAGAAAGCGAAAG-3′
1.2方法
1.2.1病毒的增殖与基因组的提取。病毒的增殖和基因组的提取,按参考文献[4]略作修改进行。
1.2.2TKL、TKR臂的PCR扩增。在50μL的PCR体系中含有如下成分:2×GC BufferⅡ(5mM Mg2+Puls)25μL、dNTP Mixture(2.5mM)2μL、Ps(5μM/L)3μL、Pa(5μM/L)3μL、模板2μL、LA Taq(5U/μL)1μL、ddH2O 13μL。均匀混合后迅速放入PCR仪进行以下程序:先94℃预变性3min,再94℃变性30s,55℃复性30s,72℃延伸1min进行30个循环,然后72℃延伸10min,PCR产物用1%琼脂糖凝胶电泳鉴定。
1.2.3含EGFP基因表达盒的重组转移载体的构建。用KpnⅠ/XbaⅠ同时酶切TKL和PUC19,回收相应片段,连接、转化、克隆、鉴定,得到PUC19-TKL;再用SphⅠ/XbaⅠ双酶切TKR和PUC19-TKL,回收相应片段,连接、转化、克隆、鉴定,得到PUC19-TK。依次消除PUC19-TK中的HindⅢ、EcoRⅠ和KpnⅠ等3个酶切位点,得到的质粒称为PUC19/HEK-TK。
用XbaⅠ酶切质粒PUC19/HEK-TK,回收片段用Klenow补平并去磷酸化;同时用SspⅠ/StuⅠ酶切质粒pcDNA3.0-EGFP,回收大小为2.9kb含CMV启动子的EGFP片段,用Klenow补平,将两片段连接、转化、克隆、鉴定,得到重组转移载体PUC19TK-EGFP。
1.2.4重组病毒的构建、筛选、纯化及鉴定。
(1)共转染。将重组转移载体PUC19TK-EGFP和PRV Ra的基因组用脂质体共转染法按Lipofectamine 2000 说明书方法转染BHK细胞,待细胞出现80%病变时收获病毒。
(2)重组病毒的筛选及纯化。将收获的病毒反复冻融3次后10倍系列稀释,接种于BHK细胞已长成单层的6孔细胞培养板,37℃吸附1h,用PBS洗涤3次,每孔加入2mL含2%琼脂糖、3%小牛血清的无酚红DMEM覆盖,置于37℃ 5% CO2培养箱中,24h后,再覆盖1.5mL含1/15 000中性红和2%琼脂糖的无酚红DMEM,于37℃ 5%CO2培养至噬斑出现,刮下发荧光的单个分散的噬斑,与BHK细胞同步接种于24孔板中进行增殖,待细胞出现80%以上病变时收获病毒。如此反复纯化3次。
(3)重组病毒的PCR鉴定。①重组病毒TK缺失基因的鉴定。将筛选到的病毒扩大培养后提取基因组,用Ps、Pa引物扩增TK缺失基因,同时用原始毒株基因组作对照。PCR产物用1%琼脂糖凝胶电泳鉴定。②重组病毒EGFP基因的鉴定。将筛选到的病毒扩大培养后提取基因组,用Ps-EGFP、Pa-EGFP引物扩增EGFP缺失基因片段。PCR产物用1%琼脂糖凝胶电泳鉴定。
2结果与分析
2.1TK基因重组左右臂的PCR扩增
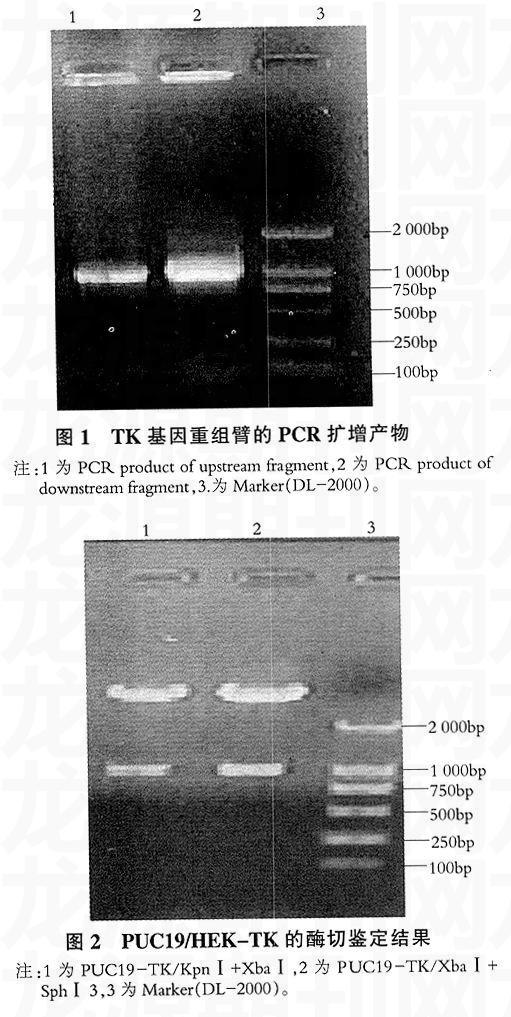
以PRV Ra株基因组为模板,用设计的引物进行PCR扩增,在1%的琼脂糖凝胶中电泳,左臂可见扩增出大小约1 000bp的片段,与预期结果相符(见图1),测序结果略。
2.2重组转移载体的鉴定
利用设计在TKL和TKR基因片段上的酶切位点对载体PUC19-TK分别进行双酶切,均得到约1 000bp的片段,与预期相符(见图2)。位于通用转移载体PUC19-EGFP多克隆位点中的EcoRⅠ为单一酶切位点,酶切后成1条带,大小约为7 600bp,与预期相符(见图3)。
2.3重组病毒的纯化及PCR鉴定
转染产物接种6孔板后,约培养36h后出现空斑,在倒置荧光显微镜下可以观察到部分病毒噬斑发出强烈的绿色荧光,说明EGFP基因在重组的PRV中获得表达,从而证实所构建的转移载体是可行的。用原始毒株和经过3次纯化的重组病毒的基因组DNA为模板,用Ps、Pa检测引物扩增TK缺失片段,原始毒株得到627bp的片段,而重组病毒株为阴性,证实已得到均一的重组病毒(见图4)。
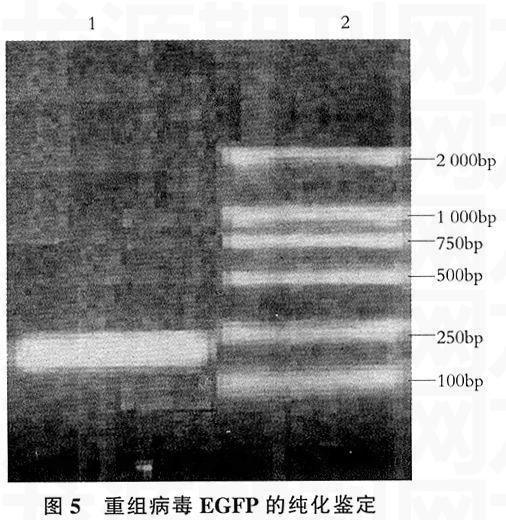
用重组病毒的基因组DNA为模板,Ps-EGFP、Pa-EGFP为引物PCR扩增EGFP基因中2 047~2 253之间长206bp的片段,结果与预期一致,表明EGFP基因确实已经插入到PRV-Ra株的基因组中(见图5)。
3讨论
TK基因是伪狂犬病病毒的主要毒力基因,它与病毒在细胞中的增殖无关,TK基因缺失后的病毒同野毒株一样在细胞培养上增殖良好,但毒力和潜伏感染的能力大大降低[5]。本研究以PRV TK基因为基础,通过扩增其同源重组左右臂,缺失203bp的基因片段,这一缺失涉及到PRV中较为保守的序列,因此将影响到TK的功能,从而降低病毒毒力。
通过同源重组来产生重组病毒是一种广泛使用的构建重组病毒的方法,但如果没有有效的筛选标记,工作量将非常大,要获得重组病毒将是非常困难的。为了克服这一问题,本研究将一种全新的报告基因——增强型绿色荧光蛋白EGFP基因插入载体PUC19/HEK-TK中用以筛选重组病毒。EGFP基因可实时表达,无需另外增加底物或辅助因子协作指示,如果病毒发生重组,在倒置荧光显微镜下就可以观察到明亮的绿色荧光,大大简化了筛选过程。
本试验用酶切的方法将带有pcDNA 3.0整套真核表达元件(CMV启动子、多克隆位点区、BGH的Poly A信号序列)的基因的片段切下来,用平末端连接的方法插入到PUC19/HEK-TK载体中,得到转移载体PUC19TK-EGFP。这样控制外源基因表达的启动子是CMV,CMV是一个适于在哺乳动物细胞中表达的高效启动子。试验也表明,将转移载体与病毒基因组共转染BHK细胞后,绿色荧光蛋白EGFP基因在CMV启动子的驱动下获得了稳定表达,同时也证实该通用转移载体的有效性,这为下一步构建表达其他病毒的重组疫苗提供了理论基础。
本试验通过酶切、补平的方法消除了pUC19中的Hind Ⅲ/EcoRⅠ/KpnⅠ等3个酶切位点,使EGFP表达盒中引入了1个常用的限制性内切酶的多克隆位点,构成通用转移载体PUC19-TK/EGFP,将来可以用1个或多个外源基因置换EGFP基因,进行多基因重组,从而产生“一针防治几病”的多价基因工程苗。
4参考文献
[1] 殷震,刘景华. 动物病毒学(第二版)[M].北京:科学出版社,1997.
[2] 钱平,李祥敏,陈焕春,等.伪狂犬病毒通用转移载体的构建及应用[J].中国预防兽医学报,2003,25(1):16-20.
[3] 方六荣,周复春,陈焕春,等.伪狂犬病毒鄂A株TK-/LacZ+突变株的构建[J].畜牧兽医学报,2001,32(3):244-248.
[4] 周复春.伪狂犬病基因缺失苗的构建[D].武汉:华中农业大学,1998.
[5] 陈焕春,周复春,方六荣,等.伪狂犬病病毒鄂A株TK-/gG-/LacZ+突变株的构建[J].病毒学报,2001,17(1):69-74.
